
Abstract
The ovine MHC class IIa is known to consist of six to eight loci located in close proximity on chromosome 20, forming haplotypes that are typically inherited without recombination. Here, we characterise the class IIa haplotypes within the Soay sheep (Ovis aries) on St. Kilda to assess the diversity present within this unmanaged island population. We used a stepwise sequence-based genotyping strategy to identify alleles at seven polymorphic MHC class IIa loci in a sample of 118 Soay sheep from four cohorts spanning 15 years of the long-term study on St. Kilda. DRB1, the most polymorphic MHC class II locus, was characterised first in all 118 sheep and identified six alleles. Using DRB1 homozygous animals, the DQA (DQA1, DQA2 and DQA2-like) and DQB (DQB1, DQB2 and DQB2-like) loci were sequenced, revealing eight haplotypes. Both DQ1/DQ2 and DQ2/DQ2-like haplotype configurations were identified and a single haplotype carrying three DQB alleles. A test sample of 94 further individuals typed at the DRB1 and DQA loci found no exceptions to the eight identified haplotypes and a haplotype homozygosity of 21.3%. We found evidence of historic positive selection at DRB1, DQA and DQB. The limited variation at MHC class IIa loci in Soay sheep enabled haplotype characterisation but showed that no single locus could capture the full extent of the expressed variation in the region.
Similar content being viewed by others

Introduction
The major histocompatibility complex (MHC) is a genomic region containing highly polymorphic genes which encode cell surface proteins involved in the presentation of pathogen-derived peptides to T cells, enabling an immune response (Klein 1986). The highly complex and polymorphic nature of the MHC region has made it a focus of many studies in immunology and evolution; however, these features make it difficult to develop locus-specific assays to genotype individual loci. Over evolutionary time, mammalian MHC loci can be viewed as going through a birth and death process (Nei et al. 1997). New MHC loci are thought to be created through gene duplication events, resulting in multiple loci harbouring similar alleles. Some are lost through decay, producing pseudogenes and gene fragments, which are characteristic of mammalian MHC regions. Additionally, allelic diversity within many MHC loci is high, and selection may favour the maintenance of numerous and divergent alleles within a population (Wakeland et al. 1990; Lenz 2011). Genotyping the MHC region is therefore often particularly challenging, and locus-specific assays can difficult to develop, as multiple loci and pseudogenes may co-amplify with primers that are too generic, whilst allelic dropout may occur with primers that are too specific (Babik et al. 2009).
The MHC class II loci exhibit high linkage disequilibrium (LD) (Lee et al. 2012), and many studies take advantage of this by genotyping a single locus, assuming that it is representative of the full haplotypic diversity (e.g. Babik et al. 2005; Harf and Sommer 2005; Bollmer et al. 2007; Biedrzycka et al. 2011; Gelasakis et al. 2013; Kamath et al. 2014). However, despite high LD, selection favouring particular alleles could mean that some alleles at specific loci are identical across different haplotypes (de Bakker et al. 2006; Traherne et al. 2006). For example, in sheep, an allele at one locus can be found in combination with multiple alleles at other loci (Hickford et al. 2007; Ballingall et al. 2015; Ali et al. 2016). Genotyping only a single locus may disguise variation at other loci, reducing the power to detect differing selection pressures. This highlights the need to characterise haplotypic variation when studying the MHC.
The unmanaged but intensively studied Soay sheep (O. aries) population on the island of Hirta, in the St. Kilda archipelago, Scotland, presents an excellent opportunity to study MHC selection in a large mammal. The Hirta population of Soay sheep originated from 107 animals which were translocated from the neighbouring smaller island of Soay in 1932 (Clutton-Brock et al. 2004), and the population has remained closed ever since. MHC variation within the study population was therefore expected to be limited compared with larger populations experiencing immigration. Since 1985, the long-term study of the Soay sheep has collected DNA samples, as well as life history and phenotype data, for many thousands of individuals (Clutton-Brock et al. 2004). Fully characterising the MHC class IIa haplotype variation within the Soay population might, therefore, be feasible.
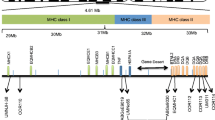
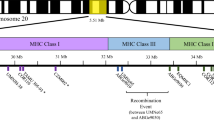
The two major families of antigen-presenting MHC molecules are class I, which present endogenous antigens (primarily from intracellular pathogens) to CD8+ T cells, and class II which present exogenous antigens (primarily from extracellular pathogens) to CD4+ T cells (Klein 1986). Unusually, the class II region within ruminants is split into two distinct subregions, class IIa and IIb (Andersson et al. 1988; van Eijk et al. 1995), with the classical class II loci, which have previously been associated with parasite resistance (see Lee et al. 2011), clustered in the class IIa region. The class IIa loci include the highly polymorphic DRB1, DQA and DQB loci, as well as the less polymorphic DRA (Ballingall et al. 2010). Duplicated pairs of DQA and DQB loci have been identified in domestic sheep, Ovis aries (Scott et al. 1987; Wright and Ballingall 1994; Ballingall et al. 2015, 2018b). Three types of DQA alleles (DQA1, DQA2 and DQA2-like; Ballingall et al. 2015) and DQB alleles (DQB1, DQB2 and DQB2-like; Ballingall et al. 2018a, b) have been identified. DQA1 and DQA2 are known to be different loci, as are DQB1 and DQB2; however, the origins of the DQA2-like and DQB2-like alleles are less well defined. Typically, DQA2-like and DQB2-like alleles are found on haplotypes in conjunction with DQA2 and DQB2, and the DQA1 and DQB1 loci are absent. Therefore, the two typical haplotype configurations are DQA1 + DQA2 with DQB1 + DQB2 and DQA2 + DQA2-like with DQB2 + DQB2-like. Whether the DQA2-like and DQB2-like alleles represent independent loci or are simply divergent alleles at the DQA1 and DQB1 loci remains unclear (Ballingall et al. 2015, 2018b). A recent study by Ali et al. (2016) identified haplotypes with all three allele types, which would suggest that the DQA2-like and DQB2-like alleles are derived from independent loci.
A previous study of MHC variation in Soay sheep using the OLADRB microsatellite located in the second intron of the class IIa locus DRB1 found evidence for selection acting on this region (Paterson et al. 1998). However, as outlined above, how well the single OLADRB microsatellite locus represents diversity across the MHC class IIa region of the Soay sheep is unknown. Since the Paterson et al. (1998) study, single locus genotyping methods targeting the polymorphic regions of the classical class IIa loci have been developed for domestic sheep. These include DRB1 (Ballingall and Tassi 2010), DQA (Ballingall et al. 2015) and DQB (Ballingall et al. 2018b). In this study, we aim to (1) genotype a sample of Soay sheep at the classical DRB1 and DQ loci using sequence-based genotyping and (2) define the MHC class IIa haplotypes in the Soay sheep study population using animals identified as homozygous for each of the DRB1 alleles. Additionally, we aim to (3) look for evidence of positive selection acting on ovine MHC alleles during their evolutionary history. Characterising the MHC class IIa loci in this population will facilitate the development of a method to determine haplotypes for large numbers of individuals, enabling subsequent investigation of the evolutionary mechanism maintaining diversity within this region.
Methods
Study system
Monitoring of Soay sheep in the Village Bay area on Hirta has been carried out intensively since 1985 (Clutton-Brock et al. 2004), including catching lambs in spring for weighing, ear-tagging and sampling for genetic analysis. Most sheep are also caught in August, when phenotypic measurements, faecal samples for strongyle egg counts and blood samples are taken. During the August catches of 2012 to 2014, aliquots of blood were collected into Tempus™ Blood RNA Tubes (ThermoFisher Scientific).
Genomic DNA preparation
Genomic DNA (gDNA) was previously extracted from either peripheral blood or ear punch tissues using either the phenol/chloroform method (Bancroft et al. 1995) or QIAGEN DNeasy or QIAamp DNA Mini kits (QIAGEN, Dusseldorf, Germany) following the manufacturer’s protocol.
Sequence-based genotyping
DRB1 genotyping
The DRB1 locus is the best characterised MHC class II locus in O. aries, and the Immuno Polymorphism Database (IPD-MHC - https://www.ebi.ac.uk/ipd/mhc) contains over 100 ovine DRB1 alleles and corresponding allelic nomenclature. Locus-specific primers and a sequence based genotyping method, which targets the polymorphic second exon, have previously been developed (Ballingall and Tassi 2010). The primer pair 330_F and 329_R (Ballingall and Tassi 2010) was tested initially as it generates full DRB1 exon 2 sequences (Table 1). However, a 1-bp deletion in the DRB1*13:01 allele generated a mixed sequence from which the alleles could not be unambiguously identified. Therefore, the forward primer 455_F, a modification described in Corbishley et al. (2016) which sits downstream of the deletion, was used in preference (Table 1). Between 27 and 31 sheep were randomly selected from each of four cohorts (1993, 1998, 2003, 2008) and genotyped to identify DRB1 homozygous individuals for subsequent analysis of DQ diversity, with the expectation that DRB1 homozygotes were more likely to be homozygous at DQ loci due to linkage disequilibrium.
DQA genotyping
For each DRB1 allele identified, four DRB1 homozygous individuals were genotyped at the DQA loci (DQA1, DQA2 and DQA2-like). Individuals homozygous for the rare allele DRB1*10:01 were not identified from the initial DRB1 screen, and thus, DQA haplotypes were determined from heterozygous individuals. Primers DQA1_F and DQA1_R were used to amplify DQA1 and primers DQA2_F and DQA2_R were used to amplify DQA2 and DQA2-like (Table 1). For DQA2/DQA2-like homozygous individuals, the DQA1 primers were not expected to generate a PCR product and the DQA2 primers were expected to amplify both DQA2 and DQA2-like loci and therefore generate a product that was heterozygous in appearance.
DQB genotyping
DQB loci (DQB1, DQB2 and DQB2-like) were characterised in individuals which were both DRB1 and DQA homozygous. Primers DQB-F and DQB1-R or DQB2-R were used to amplify the DQB loci (Table 1). The DQB primers were not completely locus-specific and some cross-amplification was expected, depending upon the alleles present (Ballingall et al. 2018b). Due to the cross-amplification of loci, it was not possible to unambiguously identify DQB alleles from DRB1*10:01 heterozygotes. A subsequent screen of a much larger number of individuals (data not included here) was necessary to find individuals homozygous for the DRB1*10:01 allele, and the corresponding DQB alleles were identified from these homozygotes.
PCR amplification
PCR reactions were carried out in a final volume of 25 μL and contained 12.5 μL Promega GoTaq Green mastermix, 0.5 μM of each primer, approximately 25 ng genomic DNA and water. Cycling conditions were 94 °C for 5 min, then 35 cycles of 94 °C for 30 or 60 s, 55–58 °C for 30 or 60 s and 72 °C for 30 or 60 s (see Table 1) and a final extension of 72 °C for 5 min. PCR products were checked using gel electrophoresis on a 1% agarose gel.
RT-PCR amplification of full length DQ transcripts
Full-length transcripts were amplified from RNA to validate the previously unidentified sequences from genomic DNA. For individuals for which Tempus™ Blood RNA Tubes were available (see Study system), DRB1 and DQA loci were amplified from genomic DNA to identify homozygous individuals for RNA extraction. Total RNA was extracted from 3 mL of Tempus tube blood stored at − 20 °C using the Tempus™ Spin RNA Isolation Kit (ThermoFisher Scientific) at half the recommended volumes. Reverse Transcription was carried out using the ImProm-II™ kit (Promega) with oligo(dT)15 primers. RT-PCR reactions were carried out using the primer combinations described in Table 1. No single primer set amplified all alleles. As RNA was not available for any individuals homozygous for haplotype H, F/H heterozygous individuals were used instead.
Cloning of multi-allelic PCR products
In order to phase novel alleles from heterozygous or multi-locus amplifications, PCR fragments were cloned into the pGEM-T easy vector (Promega). The presence of the correct insert was confirmed using colony PCR and plasmid DNA from 12 to 20 colonies was purified for sequencing using QIAprep Spin Miniprep Kit (Qiagen).
Sequencing and sequence analysis
PCR products were purified using exonuclease I and Antarctic phosphatase, except DQB2 PCR products which were gel-purified using the Macherey-Nagel Nucleospin Gel and PCR Clean Up kit, prior to Sanger sequencing. Alleles were sequenced using BigDye 3.1 chemistries on an AB 3730 genetic analyser.
Sequence analysis was carried out in Geneious 7.1.9. For DRB1 sequences, heterozygous peaks were called using the Heterozygote Plugin and checked by eye. DRB1 sequences were then compared to alleles from the IPD-MHC database using a custom BLAST. DRB1 genotypes were called when known alleles accounted for all variants within the sequence.
DQA and DQB sequences were also analysed using the Heterozygote Plugin within Geneious 7.1.9 to call heterozygous peaks. Due to multi-locus amplification, some sites contained three or four peaks, which were called by eye. A custom BLAST was used to compare sequences to an appropriate DQA database (Ballingall et al. 2015) or DQB database (Ballingall et al. 2018b). Genotypes were only called when all variants were accounted for by known alleles. Following cloning and RT-PCR of unknown alleles, the custom BLAST was updated to include the new alleles, and uncalled sequences were then compared to the updated database.
Nomenclature
The novel full-length DQA and DQB alleles identified here were named according to the nomenclature system described in Ballingall et al. (2015, 2018), were submitted to the European Nucleotide Archive (ENA; accession numbers LR025203-LR025213) and were included in the IPD-MHC database (Ballingall et al. 2018a). Genomic fragments that were considered too short to receive official nomenclature, i.e. those for which full-length transcripts could not be obtained, fell into one of two categories. Either, they matched an existing allele in GenBank that is also too short to receive official nomenclature, in which case, they were named after the accession number of the existing allele. Alternatively, the allele does not have a match in GenBank and was named according to the locus and haplotype designation and submitted to the ENA (accession numbers LR025788–LR025790).
Phylogeny of DQB
Due to the lack of locus-specific DQB primers, loci could not be determined for all amplified alleles, and thus, a phylogeny of the DQB sequences was generated to help clarify their DQB locus of origin. The DQB alleles sequenced were aligned using Clustal Omega (Sievers et al. 2011), along with previously identified DQB sequences from O. aries (Ballingall et al. 2018b), with domestic pig (accession NM_001113694.1.1) and human (accession M24364.1) DQB sequences as outgroups. Model selection for MrBayes, implemented in Topali v2 (Milne et al. 2009), selected the K80 model of DNA substitution (Kimura 1980) with gamma distribution. The phylogeny was generated in Geneious v7.1.9 using MrBayes 3.2.6 (Huelsenbeck and Ronquist 2001) using 1,000,000 generations, with a burn-in of 25,000 generations.
Validation of haplotypes
To confirm that the haplotypes characterised in homozygous individuals were consistent, DRB1 and DQA loci were genotyped in an additional 95 sheep previously not genotyped by this study. These sheep were not first order relatives of each other and were born between 1984 and 2010. These individuals were selected as they were genotyped on the Ovine Infinium HD SNP BeadChip for a previous study (Johnston et al. 2016). These individuals represent the maximum genetic variation within the population for the given number of individuals (Johnston et al. 2016).
Positive selection analyses
Positive selection acting over the evolutionary history of MHC alleles can be detected by comparing the ratio of non-synonymous (dN) to synonymous (dS) substitutions across sites (dN/dS) (Hill and Hastie 1987; Hughes and Nei 1988) and was assessed using CODEML implemented within the PAML4 package (Yang 2007). Analyses were carried out for DRB1 and for the duplicated DQ loci as all DQA loci combined (DQA1, DQA2, DQA2-like) or all DQB loci combined (DQB1, DQB2, DQB2-like) in order to maximise allelic sample sizes. As selection may differ between the duplicated loci, analyses were carried out separately for DQA1, DQA2, DQB1 and DQB2, although not for DQA2-like and DQB2-like because allele sample sizes were too small (n = 3 and n = 4, respectively). Alleles included in positive selection analyses are listed in Supplementary Table 2.
CodeML input neighbour-joining trees for each locus as described above were produced in Geneious v7.1.9 using HKY model and 5000 bootstrap replicates using O. aries alleles with assigned nomenclature (https://www.ebi.ac.uk/ipd/mhc; Ballingall et al. 2011, 2018a). The null hypothesis that neutrality is operating was first modelled for each locus using M1a in PAML, which assumes two possible classes of selection, ω0 < 1 as estimated from the data and ω1 = 1. This was then compared to the alternative hypothesis using model M2a in which positive selection is assessed using the two site classes considered under M1a as well as a third, ω2 > 1 as estimated from the data. The model which best fit the data was inferred by calculating the likelihood ratio test (LRT) p value calculated as two times the difference between the models, compared to the chi-squared distribution with two degrees of freedom (Yang 2007; Posada 2009). Type I errors can be inflated when using the LRT with high levels of recombination, so the models M7 and M8 were also tested in PAML as they have previously been shown to be more robust to recombination (Anisimova et al. 2003). Model M7 is the null model of neutrality with a β distribution, and M8 as the alternative hypothesis, with β and ω. Codon frequency model 2 (F3x4) was used throughout. Additionally, positive site selection analyses were carried out within CodeML (Bayes Empirical Bayes (BEB) within model M2a) and within HyPhy using FEL and MEME methods.
Results
Sequence-based genotyping
DRB1
Six DRB1 alleles were identified among the 118 individuals from the four cohorts born in 1993, 1998, 2003 and 2008. All six alleles were represented in the IPD-MHC database and each allele was assigned to an individual haplotype (A–F; Table 2). Allele frequencies are shown in Supplementary Fig. 1. In total, 33 (27.5%) DRB1 homozygous sheep were identified, with five of the six DRB1 alleles represented by a minimum of three homozygous individuals. Only DRB1*10:01 was not represented in homozygote form within the 118 sheep tested.
DQA loci
DQA genotyping of DRB1 homozygous individuals and DRB1*10:01 heterozygotes provided evidence for an additional two haplotypes (G and H) (Table 2). The DRB1*01:01 and DRB1*22:01 alleles were each associated with two different DQA haplotypes. DQA1 primers failed to amplify a product in three haplotypes, (DQA1 null haplotypes, A, E and G), and a DQA2-like allele was identified in each of these haplotypes.
Six DQA alleles did not match full length sequences described in (Ballingall et al. 2015), and only one of these (DQA1*Z28420 on haplotype C) matched a sequence on GenBank (accession number Z28420). Full-length transcripts (768 bp) were generated for six of these DQA alleles (Table 2). DQA1 could not be amplified from cDNA for haplotype C and official nomenclature could not be assigned, and instead, temporary nomenclature reflects the GenBank accession number (DQA1-Z28420). Nevertheless, full exon 2 sequences were obtained for all alleles, except the final 12 bp of DQA1-Z28420 (Supplementary Fig. 2).
DQB loci
Both gDNA and cDNA DQB primer pairs exhibited varying degrees of cross-locus amplification, dependent upon the alleles present (Supplementary Table 1). The DQB2-like cDNA primers did not amplify any alleles from haplotypes known to carry a DQB2-like allele, likely due to polymorphisms at the primer binding regions but perhaps due to poor quality RNA samples.
A single allele at each of the loci matched alleles in Ballingall et al. (2018b), and a further two alleles at each of DQB1 and DQB2 matched alleles in GenBank (Table 2). The remaining alleles were novel and were assigned temporary nomenclature (Table 2). Full-length sequences for the two novel alleles on haplotypes E and H could not be generated, and full-length transcripts failed to amplify for the other five alleles in haplotypes C, D, F and H as shown in Supplementary Table 1. Three DQB alleles were identified from haplotype G, two from gDNA only and one from cDNA only (Supplementary Table 1). Full exon 2 sequences were obtained for all DQB alleles (Supplementary Fig. 3).
Phylogeny of DQB loci
Phylogenetic analysis of the DQB loci revealed three distinct clusters, corresponding to the three loci DQB1, DQB2 and DQB2-like (Fig. 1). All Soay DQB alleles are located within one of the three clusters. Alleles from each haplotype fell into two different clusters, with the exceptions that haplotype G carried two alleles that clustered within DQB2, and a third that clustered most closely with DQB2-like. Haplotype C alleles were both located within the DQB2 cluster; however, it should be noted that both alleles only include exon 2 and not the 3′ UTR where the phylogenetic signal is strongest. Haplotype C alleles were therefore designated according to the primer set with which they were amplified.

Phylogeny of DQB sequences estimated using MrBayes in Geneious. Letters in brackets indicate the Soay sheep haplotype that the allele was identified on. Numbers at branches are posterior probabilities
Validation of haplotypes
Sequence-based genotyping across DRB1, DQA and DQB loci revealed eight haplotypes (Table 2). Direct sequencing of DRB1 and DQA loci in 94 individuals did not reveal any deviations from the haplotypes identified using homozygous individuals. With the addition of the novel alleles on haplotypes C, F and H to the custom BLAST, alleles from all direct sequencing products could be determined, even from DQA2/DQA2-like heterozygous products from which four alleles co-amplified. Within these 94 individuals, 21.3% were homozygous.
Positive selection and recombination analyses
For all loci, the null model of neutrality was rejected and the models incorporating positive selection were selected (Supplementary Table 3). Models M2a and M8 (positive selection) both fit the data significantly better than M1a and M7 (neutrality). However, after applying Bonferroni correction for multiple tests at DQA and DQB, (α-threshold becomes 0.017), the M1a and M7 models of neutrality are a better fit at DQA2. Additionally, positively selected sites were identified at all loci (Fig. 2), with 12 sites identified at DRB1, 14 at DQA and 13 at DQB, although only 10, 5 and 6 sites, respectively, were detected by more than one method. The majority of PSS at all three regions, DRB1, DQA and DQB, have also been identified as antigen binding sites in human homologues. Results of separate PSS analyses for DQA1 and DQA2, as well as DQB1 and DQB2, are shown in Supplementary Fig. 4 and found fewer PSS than when loci were combined with fewer overlapping PSS at DQA1/DQA2 than for DQB1/DQB2.

PSS for exon 2 of DRB1, DQA and DQB identified using BEB in CodeML, and FEL and MEME in HyPhy, where a coloured block indicates the codon was significant at > 0.95 probability (CodeML) or p < 0.05 (HyPhy). Sites denoted by asterisk indicate this position was identified as an antigen biding site within the human orthologue (Brown et al. 1993; Stern and Wiley 1994; Reche and Reinherz 2003; Bondinas et al. 2007)
Discussion
Six DRB1 alleles in Soay sheep were identified, which matched sequences previously identified in commercial Scottish sheep breeds and held in the IPD-MHC database. The level of homozygosity at the DRB1 locus was 27.1% of 118 individuals, which is higher than previously observed in other breeds of sheep (Stear et al. 2005; Herrmann-Hoesing et al. 2008). However, at the MHC class IIa haplotype level, this is an over estimate of homozygosity as some individuals homozygous for DRB1*01:01 or DRB1*22:01 are heterozygous at DQA and DQB loci. The level of MHC class IIa haplotype homozygosity reduced to 21.3% in the 94 validation samples genotyped across the DRB1 and DQ loci. Note that these samples were originally selected to maximise representation of Soay sheep diversity, so it is possible they are not totally representative of the population as a whole. No dramatic change in DRB1 allele frequencies was observed over time (Supplementary Fig. 1), but sample sizes were too small to analyse temporal trends statistically. Further investigation at the population level will provide better estimates of the level of homozygosity and whether haplotype frequencies have changed over time within the Soay sheep population.
DQA and DQB genotyping revealed a total of eight MHC class IIa haplotypes in the Soay sheep. DRB1 and DQA genotyping from MHC class IIa homozygous animals was relatively straightforward due to locus-specific primers (although the DQA2 primer pair did co-amplify DQA2-like alleles). DQB primer pairs, however, showed varying levels of cross-amplification depending upon the alleles carried by the haplotype, which made phasing of alleles challenging without extensive cloning. The limited amount of MHC class IIa haplotype diversity has made identifying variation at the DQA and DQB loci from homozygous animals possible. Furthermore, assigning DQB alleles to loci was only possible using phylogenetic analysis. DQB genotyping would be improved by further development of locus-specific primers, although this is challenging due to limited genomic sequence in this region and the lack of locus-specific characteristics in exon 2 (van Oorschot et al. 1994; Wright and Ballingall 1994). Both known DQ haplotype configurations (DQ1 + DQ2 and DQ2 + DQ2-like) were identified, as well as a novel configuration (two DQB2 alleles with DQA2 + DQA2-like + DQB2-like on haplotype G) in Soay sheep. Both DQB2 alleles on haplotype G were phylogenetically clustered within the DQB2 group (Fig. 1) and feature DQB2 locus-specific 3′ UTR sequences. The two DQB2 alleles were amplified from either cDNA (DQB2*12:01) or gDNA (DQB2*09:01). Whilst this might suggest that the DQB2*09:01 allele is not transcribed, many of the DQB alleles identified from Soay sheep failed to amplify from cDNA, including both DQB2-like alleles. Previous work has shown that all DQA and DQB loci can be expressed (Ballingall et al. 2018b), including DQB2-like*01:01 which could not be recovered from cDNA here. It is possible that the alleles we were unable to retrieve from cDNA were not expressed or were expressed at undetectable levels within the peripheral blood samples tested or in the haplotypic combinations analysed, although it perhaps more likely reflects a combination of low cDNA quality and diversity in the primer binding sites. Whether the DQB2*09:01 allele is transcribed or not remains uncertain, but it cannot be ruled out that haplotype G carries three DQB loci. A previous study of MHC class IIa haplotypes using genomic DNA in Texel sheep identified a haplotype with three DQA and three DQB loci and another with two DQA and three DQB loci (Ali et al. 2016). Non-specific transcript analysis, such as RNAseq, would be valuable is assessing if all loci and alleles are transcribed. However, variation in the number of DQ loci and DQ haplotype configurations may be more common than previously thought in O. aries.
An unusual allele was identified on haplotype A which was identical to the DQB-E1 sequence described by Herrmann-Hoessing (Unpublished, Accession number HQ728697.1) and described here as DQB2-like*03:01. This sequence, clustered with DQB2-like alleles, did not carry the single codon deletion within the second exon described in other DQB2-like alleles (Ballingall et al. 2018a, b) but does feature the typical DQB2-like 3’ UTR sequence (see Ballingall et al. 2018a). As haplotype A carries the DQA2 + DQA2-like configuration, it would be expected that it also carries the DQB2 + DQB2-like configuration. Allele DQB2-like*03:01 appears, therefore, to be a divergent DQB2-like allele. Recent developments in long read sequencing technologies, such as Oxford Nanopore or PacBio, would be valuable in determining which alleles are separate loci and which are divergent alleles, though high-quality, long-stranded DNA might be more easily obtained from domestic sheep not living on an isolated island.
Numerous alleles were shared among the MHC class IIa haplotypes within the Soay sheep. Pairwise alignment of the predicted amino acid sequences of second exons of the DRB1, DQA and DQB loci within each haplotype identified the greatest overall amino acid similarity between haplotypes A, G and A, E. However, the functional similarity of alleles is difficult to assess without detailed information on the diversity of peptides presented by each MHC molecule.
Variation in MHC class II DQ molecules is formed from the combination of α and β chains. DQ molecules are therefore the products of polymorphic DQA1 + DQB1 and DQA2 + DQB2 genes (Ballingall et al. 2018b). DQA2-like + DQB2-like combinations are likely to provide additional functional diversity as this combination has been shown to express at the cell surface following co-transfection (Ballingall et al. 2018b). Haplotypes E and G share the same DQA2*01:02:01 + DQA2-like*01:01:01 + DQB2-like*01:01:01 allelic combination, so both haplotypes will generate the same DQ2-like molecule. Therefore, an individual homozygous or heterozygous for haplotypes E and G might have fewer DQ molecules compared to other haplotype combinations. On the other hand, haplotype G carries two DQB2 genes. If both are expressed and capable of forming functional molecules in combination with DQA2*01:02:01, it would have increased DQ molecule diversity. Intra-haplotype pairing of different DQA and DQB gene combinations may also provide additional class II molecules; however, not all allelic combinations are necessarily capable of generating functional molecules (Ballingall et al. 2018b).
The eight haplotypes characterised here are likely to be representative of all MHC class IIa variation in the Soay sheep. The DRA locus, which shows only limited allelic diversity in O. aries (Ballingall et al. 2010), was not genotyped here. It is unlikely that genotyping the DRA locus would have further subdivided any haplotypes. Whilst the haplotypes from only a small number of primarily homozygous animals were characterised, the haplotypes were validated at the DRB1 and DQA loci in an additional 95 animals, and no new allelic combinations were identified. The DQB loci remain to be validated, and potential variation in the number of loci means that there is an unknown fraction of missing variation (Babik 2010). It is not certain that every haplotype has been detected; however, the extensive sequencing of multiple loci carried out throughout this study means that only alleles that occur at very low frequency will have been missed from our analysis.
Across all loci tested here, positive selection was found to be a more likely explanation for the observed diversity than neutrality, even comparing models M7 and M8 which are considered to be relatively robust to recombination (Anisimova et al. 2003). The strength of the signal was weakest at DQA2 (M2a and M8 p = 0.027), which is no longer significant after applying Bonferroni correction. Positively selected sites were also identified at all loci. Multiple PSS algorithms were tested due to their differing abilities to detect PSS under various selection scenarios, and as predicted, sites were not consistently detected by the three methodologies. The greatest variability was found at DQA and DQB (Fig. 2), which was not resolved by analysing the duplicated loci separately (Supplementary Fig. 4). This may be due to the complex nature of selection acting on the MHC, which could vary in space or time according to the parasite communities a population is exposed to, and therefore, different antigen-binding sites could be expected to experience different levels of continuous or episodic positive selection which are best detected using different algorithms. Recombination is unlikely to have a strong effect on PSS analyses (Anisimova et al. 2003). Nevertheless, there was a high level of congruence between PSS and antigen-binding sites in human homologues. This strongly suggests that there is a strong signal of positive selection within ovine MHC class IIa exon 2 loci, which is known to contain the antigen binding sites.
Whilst the DRB1 locus remains the most polymorphic ovine MHC class II locus (106 DRB1 alleles, compared to 27 DQA and 34 DQB alleles), by characterising the haplotypes within the Soay sheep, we are able to identify extensive variation at each of the DQ loci. The limited number of known alleles at the DQ loci compared to DRB1 is due to the development of effective locus-specific primers for DRB1 (Ballingall and Tassi 2010), whilst genotyping DQ allelic diversity has proved more challenging due to gene duplication and haplotype diversity. In contrast to the ovine DR molecule which is formed from the highly polymorphic DRB1 and the relatively non-polymorphic DRA (Ballingall et al. 2010), at least two ovine DQ molecules are formed from combinations of the polymorphic and duplicated DQA and DQB loci (Scott et al. 1987; Wright and Ballingall 1994; Ballingall et al. 2015, 2018b). Additionally, diversity within the DQ molecules may be achieved by combining the products of two DQA loci with two DQB loci, although not all combinations were able to produce detectable MHC class II molecules on the surface of transfected cells (Ballingall et al. 2018b). The allelic diversity observed at the ovine DQ loci suggests that it is likely to be important in immune recognition of pathogens and the loci subject to corresponding selection pressures. Without haplotype characterisation, the full complement of MHC class IIa variation cannot be detected, which may limit the ability of subsequent evolutionary analyses to detect relationships between genotypes, phenotypes and fitness and therefore elucidate the evolutionarly mechanisms maintaining the high levels of diversity observed at the ovine MHC class IIa region.
Eight MHC class IIa haplotypes have been identified in the Soay sheep population through characterisation of allelic diversity at the DRB1, DQA and DQB genes. Additionally, PSS were identified at all three gene regions, which are likely to be involved in antigen-binding given their correspondence to antigen-binding sites in human and murine homologues. The diversity observed within the Soay sheep population is reduced compared to studies of domestic sheep (Ali et al. 2016; Koutsogiannouli et al. 2016), and, using this in-depth knowledge of the MHC class IIa haplotype diversity within the Soay sheep population, it should now be possible to develop a rapid SNP-based genotyping system in order to generate large-scale population data across the more than 30-year study of Soay sheep on St. Kilda. This would greatly facilitate analyses of evolutionary processes underlying the maintenance of the variation in the MHC class IIa region.
References
Ali AOA, Stear A, Fairlie-Clarke K, Brujeni GN, Isa NMM, Salisi MSB, Donskow-Łysoniewska K, Groth D, Buitkamp J, Stear MJ (2016) The genetic architecture of the MHC class II region in British Texel sheep. Immunogenetics 69:1–7. https://doi.org/10.1007/s00251-016-0962-6
Andersson L, Lundén A, Sigurdardottir S et al (1988) Linkage relationships in the bovine MHC region. High recombination frequency between class II subregions. Immunogenetics 27:273–280. https://doi.org/10.1007/BF00376122
Anisimova M, Nielsen R, Yang Z (2003) Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164:1229–1236
Babik W (2010) Methods for MHC genotyping in non-model vertebrates. Mol Ecol Resour 10:237–251. https://doi.org/10.1111/j.1755-0998.2009.02788.x
Babik W, Durka W, Radwan J (2005) Sequence diversity of the MHC DRB gene in the Eurasian beaver (Castor fiber). Mol Ecol 14:4249–4257. https://doi.org/10.1111/j.1365-294X.2005.02751.x
Babik W, Taberlet P, Ejsmond MJ, Radwan J (2009) New generation sequencers as a tool for genotyping of highly polymorphic multilocus MHC system. Mol Ecol Resour 9:713–719. https://doi.org/10.1111/j.1755-0998.2009.02622.x
Ballingall KT, Tassi R (2010) Sequence-based genotyping of the sheep MHC class II DRB1 locus. Immunogenetics 62:31–39. https://doi.org/10.1007/s00251-009-0410-y
Ballingall KT, Rocchi M, McKeever DJ, Wright F (2010) Trans-species polymorphism and selection in the MHC class II DRA genes of domestic sheep. PLoS One 5:e11402. https://doi.org/10.1371/journal.pone.0011402
Ballingall KT, Herrmann-Hoesing LM, Robinson J, Marsh SGE, Stear MJ (2011) A single nomenclature and associated database for alleles at the major histocompatibility complex class II DRB1 locus of sheep. Tissue Antigens 77:546–553. https://doi.org/10.1111/j.1399-0039.2011.01637.x
Ballingall KT, Steele P, Lantier I, Cotelli M, Todd H, Lopez G, Martin E, Lantier F (2015) An ancient interlocus recombination increases class II MHC DQA diversity in sheep and other Bovidae. Anim Genet 46:333–336. https://doi.org/10.1111/age.12290
Ballingall KT, Dicks K, Kyriazopoulou P, Herrmann-Hoesing L (2018a) Allelic nomenclature for the duplicated MHC class II DQ genes in sheep. Immunogenetics. https://doi.org/10.1007/s00251-018-1096-9
Ballingall KT, Lantier I, Todd H, Lantier F, Rocchi M (2018b) Structural and functional diversity arising from intra and inter haplotype combinations of duplicated DQA and B loci within the ovine MHC. Immunogenetics 70:257–269. https://doi.org/10.1007/s00251-017-1029-z
Bancroft DR, Pemberton JM, King P (1995) Extensive protein and microsatellite variability in an isolated, cyclic ungulate population. Heredity 74:326–336. https://doi.org/10.1038/hdy.1995.47
Biedrzycka A, Kloch A, Buczek M, Radwan J (2011) Major histocompatibility complex DRB genes and blood parasite loads in fragmented populations of the spotted suslik Spermophilus suslicus. Mamm Biol 76:672–677. https://doi.org/10.1016/j.mambio.2011.05.002
Bollmer JL, Vargas FH, Parker PG (2007) Low MHC variation in the endangered Galapagos penguin (Spheniscus mendiculus). Immunogenetics 59:593–602. https://doi.org/10.1007/s00251-007-0221-y
Bondinas GP, Moustakas AK, Papadopoulos GK (2007) The spectrum of HLA-DQ and HLA-DR alleles, 2006: a listing correlating sequence and structure with function. Immunogenetics 59:539–553. https://doi.org/10.1007/s00251-007-0224-8
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39. https://doi.org/10.1038/364033a0
Clutton-Brock TH, Pemberton JM, Coulson T (2004) The sheep of St Kilda. In: Clutton-Brock TH, Pemberton JM (eds) Soay sheep: dynamics and selection in an island population. Cambridge University Press, Cambridge, pp 17–50
Corbishley A, Connelley TK, Wolfson EB et al (2016) Identification of epitopes recognised by mucosal CD4+ T-cell populations from cattle experimentally colonised with Escherichia coli O157:H7. Vet Res 47(90):W289–W294. https://doi.org/10.1093/nar/gki390
de Bakker PIW, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, Morrison J, Richardson A, Walsh EC, Gao X, Galver L, Hart J, Hafler DA, Pericak-Vance M, Todd JA, Daly MJ, Trowsdale J, Wijmenga C, Vyse TJ, Beck S, Murray SS, Carrington M, Gregory S, Deloukas P, Rioux JD (2006) A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 38:1166–1172. https://doi.org/10.1038/ng1885
Gelasakis AI, Arsenos G, Hickford JGH, Zhou H, Psifidi A, Valergakis GE, Banos G (2013) Polymorphism of the MHC-DQA2 gene in the Chios dairy sheep population and its association with footrot. Livest Sci 153:56–59. https://doi.org/10.1016/j.livsci.2013.02.011
Harf R, Sommer S (2005) Association between major histocompatibility complex class II DRB alleles and parasite load in the hairy-footed gerbil, Gerbillurus paeba, in the southern Kalahari. Mol Ecol 14:85–91. https://doi.org/10.1111/j.1365-294X.2004.02402.x
Herrmann-Hoesing LM, White SN, Kappmeyer LS, Herndon DR, Knowles DP (2008) Genomic analysis of Ovis aries (Ovar) MHC class IIa loci. Immunogenetics 60:167–176. https://doi.org/10.1007/s00251-008-0275-5
Hickford JGH, Zhou H, Fang Q (2007) Haplotype analysis of the DQA genes in sheep: evidence supporting recombination between the loci. J Anim Sci 85:577–582. https://doi.org/10.2527/jas.2006-217
Hill RE, Hastie ND (1987) Accelerated evolution in the reactive centre regions of serine protease inhibitors. Nature 326:96–99. https://doi.org/10.1038/326096a0
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hughes AL, Nei M (1988) Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335:167–170. https://doi.org/10.1038/335167a0
Johnston SE, Bérénos C, Slate J, Pemberton JM (2016) Conserved genetic architecture underlying individual recombination rate variation in a wild population of Soay sheep (Ovis aries). Genetics 203:583–598
Kamath PL, Turner WC, Küsters M, Getz WM (2014) Parasite-mediated selection drives an immunogenetic trade-off in plains zebras (Equus quagga). Proc R Soc Lond B Biol Sci 281:20140077. https://doi.org/10.1098/rspb.2014.0077
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/BF01731581
Klein J (1986) The natural history of the major histocompatibility complex. Wiley, New York
Koutsogiannouli EA, Moutou KA, Stamatis C, Mamuris Z (2016) MHC class II DRB1 and DQA2 gene polymorphisms in four indigenous breeds of sheep (Ovis aries). Mamm Biol 81:628–636. https://doi.org/10.1016/j.mambio.2016.08.002
Lee CY, Munyard KA, Gregg K, Wetherall JD, Stear MJ, Groth DM (2011) The influence of MHC and immunoglobulins A and E on host resistance to gastrointestinal nematodes in sheep. J Parasitol Res 2011:101848–101811. https://doi.org/10.1155/2011/101848
Lee CY, Qin J, Munyard KA, Siva Subramaniam N, Wetherall JD, Stear MJ, Groth DM (2012) Conserved haplotype blocks within the sheep MHC and low SNP heterozygosity in the class IIa subregion. Anim Genet 43:429–437. https://doi.org/10.1111/j.1365-2052.2011.02268.x
Lenz TL (2011) Computational prediction of MHC II-antigen binding supports divergent allele advantage and explains trans-species polymorphism. Evolution 65:2380–2390. https://doi.org/10.1111/j.1558-5646.2011.01288.x
Marsh SGE, Albert ED, Bodmer WF, Bontrop RE, Dupont B, Erlich HA, Fernández-Viña M, Geraghty DE, Holdsworth R, Hurley CK, Lau M, Lee KW, Mach B, Maiers M, Mayr WR, Müller CR, Parham P, Petersdorf EW, Sasazuki T, Strominger JL, Svejgaard A, Terasaki PI, Tiercy JM, Trowsdale J (2010) Nomenclature for factors of the HLA system, 2010. Tissue Antigens 75:291–455. https://doi.org/10.1111/j.1399-0039.2010.01466.x
Milne I, Lindner D, Bayer M, Husmeier D, McGuire G, Marshall DF, Wright F (2009) TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics 25:126–127. https://doi.org/10.1093/bioinformatics/btn575
Nei M, Gu X, Sitnikova T (1997) Evolution by the birth-and-death process in multigene families of the vertebrate immune system. PNAS 94:7799–7806
Paterson S, Wilson K, Pemberton JM (1998) Major histocompatibility complex variation associated with juvenile survival and parasite resistance in a large unmanaged ungulate population. PNAS 95:3714–3719. https://doi.org/10.1073/pnas.95.7.3714
Posada D (2009) Selecting models of evolution. In: Lemey P, Salemi M, Vandamme A-M (eds) The phylogenetic handbook. Cambridge University Press, Cambridge, pp 345–361
Reche PA, Reinherz EL (2003) Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J Mol Biol 331:623–641. https://doi.org/10.1016/S0022-2836(03)00750-2
Scott PC, Choi C-L, Brandon MR (1987) Genetic organization of the ovine MHC class II region. Immunogenetics 25:116–122. https://doi.org/10.1007/BF00364277
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Stear MJ, Innocent GTT, Buitkamp J (2005) The evolution and maintenance of polymorphism in the major histocompatibility complex. Vet Immunol Immunopathol 108:53–57. https://doi.org/10.1016/j.vetimm.2005.07.005
Stern LJ, Wiley DC (1994) Antigenic peptide binding by class I and class II histocompatibility proteins. Structure 2:245–251. https://doi.org/10.1016/S0969-2126(00)00026-5
Traherne JA, Horton R, Roberts AN, Miretti MM, Hurles ME, Stewart CA, Ashurst JL, Atrazhev AM, Coggill P, Palmer S, Almeida J, Sims S, Wilming LG, Rogers J, de Jong PJ, Carrington M, Elliott JF, Sawcer S, Todd JA, Trowsdale J, Beck S (2006) Genetic analysis of completely sequenced disease-associated MHC haplotypes identifies shuffling of segments in recent human history. PLoS Genet 2:e9. https://doi.org/10.1371/journal.pgen.0020009
van Eijk MJT, Beever JE, Da Y et al (1995) Genetic mapping of BoLA-A, CYP21, DRB3, DYA, and PRL on BTA23. Mamm Genome 6:151–154. https://doi.org/10.1007/BF00303266
van Oorschot RAH, Maddox JF, Adams LA, Fabb SA (1994) Characterization and evolution of ovine MHC class II DQB sequence polymorphism. Anim Genet 25:417–424. https://doi.org/10.1111/j.1365-2052.1994.tb00532.x
Wakeland EK, Boehme S, She JX, Lu CC, McIndoe RA, Cheng I, Ye Y, Potts WK (1990) Ancestral polymorphisms of MHC class II genes: divergent allele advantage. Immunol Res 9:115–122. https://doi.org/10.1007/BF02918202
Wright H, Ballingall KT (1994) Mapping and characterization of the DQ subregion of the ovine MHC. Anim Genet 25:243–249. https://doi.org/10.1111/j.1365-2052.1994.tb00200.x
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. https://doi.org/10.1093/molbev/msm088
Acknowledgements
We thank the National Trust for Scotland and the Scottish Natural Heritage for permission to work on St. Kilda and QinetiQ for logistics and support. Thanks to Wei Huang, University of Edinburgh for sequencing haplotype D at DQB loci, and Helen Todd, Moredun Research institute for guidance with lab work.
Funding
KLD was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) (grant number BB/J01446X/1). KTB receives funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 731014KB (VetBioNet) and acknowledges the support received from the Scottish Government’s strategic research programme. The Soay Sheep Project is funded by the UK Natural Environment Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 20 kb)
Supplementary Figure 1.
Frequency of DRB1 alleles in four cohorts of Soay sheep: 1993 (white), 1998 (light grey), 2003 (dark grey) and 2008 (black). (JPEG 52 kb)
Supplementary Figure 2.
DQA exon 2 nucleotide sequences of alleles identified in Soay sheep. Nucleotides are numbered from the first base of the translation start codon in exon 1, and thus exon 2 begins at base 83. Dots (.) indicate identity to DQA1*03:01:01 and dashes (-) indicate missing sequence. (PNG 361 kb)
Supplementary Figure 3.
DQB exon 2 nucleotide sequences of alleles identified in Soay sheep. Nucleotides are numbered from the first base of the translation start codon in exon 1, and thus exon 2 begins at base 110. Dots (.) indicate identity to DQB1*02:01:01 and dashes (-) indicate missing sequence. (PNG 409 kb)
Supplementary Figure 4.
PSS for exon 2 of DQA and DQB loci analysed together and separately using BEB in CodeML, and FEL and MEME in HyPhy, where a coloured block indicates the codon was significant at >0.95 probability (CodeML) or p < 0.05 (HyPhy). Sites denoted * indicate this position was identified as an antigen biding site within the human orthologue (Brown et al. 1993; Stern and Wiley 1994; Reche and Reinherz 2003; Bondinas et al. 2007). (PNG 8 kb)
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Dicks, K.L., Pemberton, J.M. & Ballingall, K.T. Characterisation of major histocompatibility complex class IIa haplotypes in an island sheep population. Immunogenetics 71, 383–393 (2019). https://doi.org/10.1007/s00251-019-01109-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-019-01109-w