
Abstract
The fungus Stachybotrys (S.) chartarum was isolated from culinary herbs, damp building materials, and improperly stored animal forage. Two distinct chemotypes of the fungus were described that produced either high-cytotoxic macrocyclic trichothecenes (S type) or low-cytotoxic atranones (A type). Recently, two distinct gene clusters were described that were found to be necessary for the biosynthesis of either macrocyclic trichothecenes (21 SAT (Satratoxin) genes) or atranones (14 ATR (Atranone) genes). In the current study, PCR primers were designed to detect SAT and ATR genes in 19 S. chartarum chemotype S and eight S. chartarum chemotype A strains. Our analysis revealed the existence of three different genotypes: satratoxin-producing strains that harbored all SAT genes but lacked the ATR gene cluster (genotype S), non-satratoxin-producing strains that possessed the ATR genes but lacked SAT genes (genotype A), and a hitherto undescribed hybrid genotype among non-satratoxin-producing strains that harbored all ATR genes and an incomplete set of SAT genes (genotype H). In order to improve the discrimination of genotypes, a triplex PCR assay was developed and applied for the analysis of S. chartarum and S. chlorohalonata cultures. The results show that genes for macrocyclic trichothecenes and atranones are not mutually exclusive in S. chartarum. Correlation of the new genotype-based concept with mycotoxin production data shows also that macrocyclic trichothecenes are exclusively produced by S. chartarum genotype S strains.
Similar content being viewed by others
Introduction
Stachybotrys (S.) spp. were detected on dead plant materials (e.g., herbs, straw, and hay) and other cellulosic substrates (Biermaier et al. 2015; El-Kady and Moubasher 1982). Due to its high cellulolytic potential, Stachybotrys spp. were, together with a variety of other fungal genera, demonstrated to grow on building materials (e.g., wallpaper, plasterboard, or wooden lining) under high water activity conditions (Gravesen et al. 1999; Nielsen et al. 2004). A strong association was detected between Stachybotrys ssp. and gypsum materials as well as wall papers (Andersen et al. 2011). On these materials, Stachybotrys spp. were also associated with other fungi, i.e., Acremonium spp., Ulocladium spp., and Penicillium chrysogenum. S. chartarum and S. chlorohalonata were found to be the most frequently isolated species of the genus Stachybotrys (Wang et al. 2015; Lombard et al. 2016). The former species was sub-divided into two distinct chemotypes, the high-cytotoxic chemotype S and the low-cytotoxic chemotype A. Cultures of the two chemotypes were found to produce either macrocyclic trichothecenes (S type) or atranones (A type) (Jarvis et al. 1995; Andersen et al. 2003; Hinkley et al. 2000, 2003).
In the literature, S. chartarum was associated with human (Etzel 2003) and animal (Hintikka 1977a, 1977b) diseases (Johanning and Yang 1994; Nikulin et al. 1994). Stachybotryotoxicosis is a condition that was found to occur in horses, cattle, sheep, and chickens after the oral uptake of Stachybotrys-contaminated feed (Forgacs et al. 1958; Kriek and Marasas 1983; Schneider et al. 1979; Vertinsky 1940). In humans, exposure to macrocyclic trichothecenes and other metabolites of S. chartarum, e.g., stachylysin, was suspected to cause pulmonary hemorrhage in infants and other symptoms related to the sick building syndrome (SBS) (Dearborn et al. 1999, 2002; Etzel et al. 1998; Vesper et al. 2001, Johanning et al. 1999). Even though there is evidence that links this fungus and its metabolites to several types of illnesses, the causative role of Stachybotrys spp. in the etiology of the SBS has been controversially discussed in the more recent literature (Miller et al. 2003; CDC 2002).
Satratoxins and other macrocyclic trichothecenes were shown to have been produced during growth of S. chartarum on different building materials. Thus, satratoxin H was proposed as a biomarker for the presence of macrocyclic trichothecenes (Aleksic et al. 2016). Such compounds bind irreversibly to the 60S ribosomal subunit of mammalian cells and inhibit protein biosynthesis (Hernandez and Cannon 1982; Rocha et al. 2005; Ueno 1977). They represent the most cytotoxic trichothecenes currently known (Hanelt et al. 1994). Atranones A and C were found to induce significant inflammatory responses in a mouse lung model (Rand et al. 2006). However, their cytotoxic effects were much lower as compared to macrocyclic trichothecenes (Abbas et al. 2002). Production of atranones and macrocyclic trichothecenes was postulated to be mutually exclusive in strains of the respective chemotypes (Andersen et al. 2003). Strains of the A and the S chemotypes could not be differentiated by cultural and morphological features in previous studies (Andersen et al. 2003). Moreover, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis did not allow for an unequivocal distinction of the two chemotypes (Gruenwald et al. 2015; Ulrich et al. 2016). Andersen et al. (2003) demonstrated that macrocyclic trichothecene-producing strains could be differentiated from atranone producers by the presence of a stable C-to-T exchange at nucleotide position 279 in their TRI5 genes.
Comparison of the genomes of three S. chartarum strains representing both chemotypes as well as one strain of S. chlorohalonata revealed the presence of common, but also of chemotype-specific, gene clusters. These were attributed to the biosynthesis of satratoxins and atranones, respectively (Semeiks et al. 2014). Two isolates of S. chartarum (IBT 7711 and IBT 40293) produced macrocyclic trichothecenes and contained genes that were suggested to be involved in the respective biosynthetic pathway. Genes SAT1–SAT21 were organized in three SAT core gene clusters, SC1–SC3 (Semeiks et al. 2014). Moreover, two atranone-producing isolates (IBT 40288 and S. chlorohalonata IBT 40285) lacked all SAT genes but instead contained a cluster of 14 ATR genes that could have been involved in atranone biosynthesis (Semeiks et al. 2014). From their data, these authors concluded that the absence of the ATR gene cluster from the sequenced S-type strains provides evidence for the concept of mutual exclusiveness of SAT and ATR gene clusters in the two chemotypes of S. chartarum.
Based on the earlier findings, we aimed to develop a PCR assay to distinguish the two chemotypes of S. chartarum. In this context, we designed primers for the detection of each of the genes within the SAT and ATR clusters in the chemotypes of S. chartarum. Here, we report on findings resulting in the identification of a new genotype of S. chartarum. This genotype harbors both the complete ATR gene cluster and an incomplete set of SAT genes. These result in failure of such strains to produce macrocyclic trichothecenes.
Materials and methods
Chemicals
LC-MS-grade acetonitrile (ACN) and methanol were purchased from Th. Geyer (Renningen, Germany). Ultrapure water was obtained by purifying water through an UltraClear® TP UV UF TM from Evoqua Water Technologies (Barsbuettel, Germany). Formic acid and ammonium formate used as additives for LC-MS solvents were obtained from Th. Geyer (Renningen, Germany) and Fluka (Steinheim, Germany), respectively. Standards for roridin E and L-2; satratoxins F, G, and H; and verrucarin J were not commercially available, and these toxins were therefore qualitatively determined after purification of these substances from rice cultures as described earlier (Gareis and Gottschalk 2014).
Fungal cultures and culture conditions
In this study, 28 fungal strains were used (Table 1). The set of strains comprised six reference strains [available at CBS (Westerdijk Fungal Biodiversity Institute), ATCC (American Type Culture Collection), and IBT Culture Collection of Fungi, Denmark] and 22 field isolates (Culture Collection of Food Safety, Faculty of Veterinary Medicine, Ludwig-Maximilians-University, Munich, Germany, and Chair of Technical Microbiology, TUM School of Life Sciences Weihenstephan, Technical University of Munich, Freising, Germany, from which cultures can be retrieved upon request). The strains were initially characterized using previously described molecular and mass spectrometric methods (Andersen et al. 2003; Ulrich et al. 2016) and were confirmed as either S. chartarum or S. chlorohalonata. Fungal stock cultures were maintained in glycerol at − 80 °C as described by Niessen et al. (Niessen and Vogel 2010). Working cultures of Stachybotrys spp. were grown on 2% malt extract agar plates [MEA, 20 g/L malt extract, 2 g/L soy peptone, and 15 g/L agar (Difco, Heidelberg, Germany), adjust to pH 5.4]. Prior to use, all media were sterilized by autoclaving at 121 °C for 15 min. All cultures were grown at ambient temperature (AT, 22 ± 1 °C). For DNA extraction, cultures were grown in 50 mL of malt extract broth in 100-mL Erlenmeyer flasks on a horizontal shaker at 80 rpm at. Cultures were inoculated with a small piece of mycelium from working culture plates.
Isolation of fungal DNA and PCR products
Fungal mycelia were vacuum-filtered through sterile 70-mm-diameter filter discs MN 615 (Macherey-Nagel, Düren, Germany) and washed twice with sterile tap water. Extraction of DNA from 100 mg mycelia was performed using the peqGOLD Fungal DNA Mini kit (VWR International, Erlangen, Germany) according to the manufacturer’s instructions. Extraction of DNA from agarose gels was performed using the MinElute Gel Extraction kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Genomic DNA was eluted and suspended in filtered, sterile, deionized water for all extraction protocols (Filtropur S 0.2 μm, Sarstedt, Nümbrecht, Germany). DNA concentrations were determined with a NanoDrop 1000 spectrophotometer (PeQlab, Erlangen, Germany).
Primer design
Primers for the detection of each SAT and ATR gene in S. chartarum and S. chlorohalonata were designed using sequence information available from the National Center for Biotechnology Information (NCBI, Bethesda, USA) database. Genome sequences of macrocyclic trichothecene-producing S. chartarum strains IBT 40293 (scaffold 155, accession KL650302.1) and IBT 7711 (scaffold 234, accession KL647604.1) as well as the atranone-producing strain IBT 40288 (scaffold 1, accession KL659150.1) and strain S. chlorohalonata IBT 40285 (scaffold 175, accession KL659308.1; Semeiks et al. 2014) were used to extract 21 SAT and 14 ATR gene sequences. Sequence alignments were made using the BioEdit software package version 7.2.5 (Hall 1999). The web-based tool “Primer Blast” (Ye et al. 2012) available at https://www.ncbi.nlm.nih.gov/tools/primer-blast/ was used for primer design with the following settings: product size 70–1000 bp, melting temperature 59–61 °C, automatic search mode, at least 5 mismatches to unintended targets, and maximum target size 4000 bp. All other settings were used in default mode. Differences in the individual product sizes allowed differentiation of fragments in a multiplex PCR assay. A list of the primers used in this study is given in Online Resource 1.
DNA amplification and PCR product analysis
Trichodiene synthase 5 (tri5) gene amplification and sequencing
The tri5 PCR master mix contained per 50-μL reaction 5.0 μL of 10× Taq buffer, 5.0 μL MgCl2 (final concentration 0.025 mol/L), 5.0 μL dNTP (final concentration 0.002 mol/L), 12.5 μL Taq polymerase (Thermo Prime Taq DNA Polymerase, Thermo Scientific, Schwerte, Germany; final concentration 1.25 U/μL), 1 μL of each primer (tri5f/tri5r, final concentration 1 × 10−5 mol/L), and 50 μL sterile demineralized water. DNA amplification of the TRI5 gene region was performed using the following temperature protocol: melting 1 × 95 °C for 10 min followed by 40× melting at 95 °C for 60 s, annealing at 51 °C for 30 s, elongation at 72 °C for 45 s followed by 1× final elongation at 72 °C for 10 min. The TRI5 PCR product was sequenced to differentiate between S. chartarum type A and type S according to Cruse et al. (2002) and Andersen et al. (2003). Prior to sequencing, the TRI5 PCR product was purified using the QIAquick® purification kit (Qiagen, Venlo, Germany) according to the manufacturer’s instructions and diluted with sterile demineralized water to the final concentration of 5 ng/μL. Sequencing was performed by Eurofins MWG Operon (Ebersberg, Germany). The obtained nucleotide sequences were compared with the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/) and MycoBank (http://www.mycobank.org/quicksearch.aspx) (Robert et al. 2005) online databases using the BLAST tool (Altschul et al. 1990).
PCR analysis of satratoxin and atranone cluster genes
Primers used in PCRs for the detection of the SAT and ATR genes are listed in Online Resource 1. Reactions were run as multiplex PCR using between three and five pairs of primers per reaction. Primers were selected to result in a common melting temperature of 60 °C and combined to result in products with clearly distinct fragment sizes. The master mix contained per 25-μL reaction 2.5 μL of 10× Taq buffer including 0.015 mol/L MgCl2 (MP Biomedicals, Heidelberg, Germany), 0.5 μL of dNTP solution (MP Biomedicals, 0.01 mol/L, A, T, G, C each), 0.5 μL of each primer (0.05 mol/L, custom synthesis by Eurofins, Ebersberg, Germany), 0.25 μL of Taq DNA polymerase (MP Biomedicals, 5 U/μL), and sterile demineralized water up to 24 μL. Formamide (0.25 μL per reaction) was added in the triplex PCR for the combined detection of the S. chartarum genotypes and S. chlorohalonata. One microliter of isolated genomic DNA (ca. 50 ng/μL) of the tester strains was added as template before the PCRs were run in a Master Cycler gradient 96 thermal cycler (Eppendorf, Hamburg, Germany) using the following temperature protocol: melting 1× at 94 °C for 5 min followed by 35× melting at 94 °C for 60 s, annealing at 59–62 °C for annealing time as given in Online Resource 1, elongation at 72 °C for 60 s followed by 1× final extension at 72 °C for 5 min. Elongation time during regular cycles was prolonged to 90 s for products exceeding 700 bp in size. Amplification products were separated on 1.3% (w/v) LE agarose (Biozym, Hessisch Oldendorf, Germany) prepared in 1× TAE buffer (0.05 mol/L Tris (pH 7.6), 0.02 mol/L acetic acid, 0.001 mol/L EDTA). The GeneRuler 100-bp ladder (Thermo Scientific, Schwerte, Germany) was used as molecular weight marker. Gels were stained with dimidium bromide (0.5 μg/mL) and inspected on a UVT 28M UV-transilluminator (Herolab, Wiesloch, Germany). Results were documented using a digital gel documentation system (Intas Science Imaging, Göttingen, Germany) and matching GDS frame grabber software (Intas, Göttingen, Germany).
MALDI-TOF MS measurement
Mycelia of the analyzed strains were cultured and proteins were extracted according to Ulrich et al. (2016). MALDI-TOF MS measurements were performed using an Auto Flex Speed mass spectrometer (Bruker Daltoniks, Bremen, Germany) according to the Bruker Daltoniks guidelines (Bruker Daltoniks 2013). Each strain was measured in eight technical replicates, and data were processed using the Biotyper OC software (Bruker Daltoniks, Bremen, Germany). The Biotyper database (version 3.1.66) plus an inhouse database (Ulrich et al. 2016) was used for identification of strains to the species level.
LC-MS/MS analysis—extraction method
For macrocyclic trichothecene analysis, each strain was cultured on three parallel MEA plates for 21 days at 25 °C and 95% air humidity. Cultures were stored at − 20 °C until extraction. Prior to extraction, each of the three parallel plates was separately transferred to a mixing bag and 50 mL ACN/H2O (84/16, v/v) was added. Bags were treated for 5 min in a bag mixer (BagMixer 400, InterScience, St Nom la Bretèche, France). The sample extracts were filtered through a paper filter, and an aliquot of 5 mL was evaporated to dryness under a gentle flow of nitrogen at + 50 °C. The residues were suspended in 1 mL ACN/H2O (30/70, v/v) using ultrasonication (5 min) and were subsequently filtered through a polyvinylidene fluoride (PVDF) syringe filter (0.45 μm, Berrytec, Grünwald, Germany) into 1.5-mL glass sample vials.
LC-MS/MS analysis—measurement
The LC-MS/MS system consisted of an HPLC apparatus (Shimadzu LC-20AB, SIL-20AC HT, CTO-20AC, CBM-20A, Duisburg, Germany) and an API 4000 triple quadrupole mass spectrometer (Sciex, Darmstadt, Germany). The substance-specific parameters used for multiple-reaction-monitoring (MRM) measurement are listed in Online Resource 2. The following source parameters were applied: ion spray voltage (ESI+), 5500 V; temperature, 550 °C; nebulizer gas, 50 psi; heating gas, 60 psi; curtain gas, 30 psi; and collision gas (nitrogen), level 9. A Synergi™ Polar-RP (150 × 2 mm, 4 μm; Phenomenex, Aschaffenburg, Germany) was used as an analytical column protected by a guard column. The binary linear gradient consisted of eluent A (deionized water containing 5 mmol/L ammonium formate, 0.1% formic acid) and eluent B (LC-MS-grade methanol, 5 mmol/L ammonium formate, 0.1% formic acid) with a flow rate of 0.4 mL/min: 0 min 10% B, 10 min 40% B, and 26 min 100% B. The column was equilibrated at starting conditions for 6 min prior to each run. Analyst (Version 1.6.2) and MultiQuant software (Version 3.0.1), both provided by Sciex (Darmstadt, Germany), were used for data acquisition and processing.
Results and discussion
In this study, we analyzed 27 strains of S. chartarum and one strain of S. chlorohalonata. Using MALDI-TOF MS, the respective species identifications were confirmed for all strains (Table 1). Since S. chartarum strains cannot be further differentiated into chemotypes by this method (Ulrich et al. 2016), the production of macrocyclic trichothecenes was analyzed to assign chemotypes to the respective strains. According to the LC-MS/MS data, 13 of the 27 S. chartarum strains produced macrocyclic trichothecenes, whereas S. chlorohalonata and 14 other S. chartarum strains did not (Table 1). Further analysis revealed that all strains with a thymidine (T) at nucleotide position 279 of the TRI5 gene (S. chartarum type A) did not produce macrocyclic trichothecenes (Andersen et al. 2003). All strains that produced macrocyclic trichothecenes harbored a cytidine (C) at the same position (S. chartarum type S). However, the type S configuration in nucleotide position 279 was also found in six of the S. chartarum strains that produced no macrocyclic trichothecenes. The latter finding indicates that in contrast to findings by Andersen et al. (2003), the TRI5 gene sequence does not seem to be a reliable marker for the differentiation of S. chartarum strains that produce macrocyclic trichothecenes from those that do not. From the results, it can be concluded that new and reliable genetic markers are needed for a more accurate differentiation between high- and low-cytotoxic strains in S. chartarum.
Development of a triplex PCR assay for chemotype differentiation
In order to differentiate strains of S. chartarum type A from type S as well as from S. chlorohalonata, PCR primers were designed to hybridize with genes SAT19, ATR6, and ATR4, respectively, to allow for specific identification. The SAT19 gene was selected because it was supposed to be unique in type S of S. chartarum (Semeiks et al. 2014). Moreover, it showed a very high degree of sequence homology between reference strains and included only one intron. It was therefore chosen for the design of diagnostic PCR primers for the S. chartarum type S. The ATR6 gene was supposed to be unique in S. chartarum type A strains and had only weak homologies with the ATR6 sequence in S. chlorohalonata. In the current study, it was therefore used for the design of diagnostic PCR primers for S. chartarum type A strains with no cross reaction expected to occur with S. chlorohalonata. The ATR4 gene included regions of low homology between S. chartarum and S. chlorohalonata. One such region was used to design diagnostic PCR primers for S. chlorohalonata. The three primer pairs were combined in a triplex PCR resulting in three DNA fragments of different sizes for the different types and species (see Online Resource 3). S. chartarum type A was characterized by a 230-bp fragment, whereas S. chartarum type S showed a fragment of 346 bp. Amplification of S. chlorohalonata DNA resulted in two fragments of 544 bp and 230 bp, respectively. The latter result suggested that S. chlorohalonata possesses both an ATR6 gene and an ATR4 gene and that the designed primers hybridize to both of them. In order to omit the production of a 230-bp product, formamide was added to the PCR which resulted in a weaker band but did not completely prevent this cross reaction. However, since the combination of a 544-bp fragment with a 230-bp fragment only occurred in S. chlorohalonata, the primers are still useful to identify strains of that species. All in all, results showed that the species and types could be readily distinguished by applying the developed triplex PCR assay.
Screening of S. chartarum strains with triplex PCR
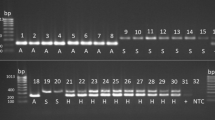
Applying the triplex PCR assay to a larger number of S. chartarum isolates revealed that not all isolates exhibited the expected banding pattern. Figure 1 displays 15 analyzed strains of which only 8 showed the banding pattern that was expected from the previously described results (see Online Resource 3). Four of the 15 S. chartarum strains produced fragments typical for type S (346 bp) together with fragments typical for type A (230 bp) (see Fig. 1, lanes 5, 10, 11, and 13). This result indicated that at least three distinct PCR patterns can be obtained from S. chartarum strains using the triplex PCR described above. Because the banding patterns were supposed to represent three different genetic constellations, three different genotypes were assigned to the corresponding patterns: genotype S (a single 346-bp fragment), genotype A (a single 230-bp fragment), and genotype H (a hybrid pattern with two fragments of 230 bp and 346 bp, respectively). Strains of S. chlorohalonata were characterized by two PCR amplicons of 544 bp and 230 bp as described above.

Triplex PCR for the combined identification of Stachybotrys (S.) chartarum type S and type A as well as S. chlorohalonata. Primers SAT19-Stype-f/r, ATR6-7Atype-f/r, and ATR4-1chloro-f/r were combined. Lanes: M = GeneRuler 100-bp ladder; lane 1 = S. chlorohalonata CBS 222.46; lane 2 = S. chlorohalonata CBS 329.37; lane 3 = S. chartarum Sp 2630; lane 4 = S. chartarum Sp 2674; lane 5 = S. chartarum Sp 2675; lane 6 = S. chartarum Sp 2676; lane 7 = S. chartarum 14/3; lane 8 = S. chartarum 95/121; lane 9 = S. chartarum D-9662; lane 10 = S. chartarum HMRF 4; lane 11 = S. chartarum HMRB 10; lane 12 = S. chartarum CBS 129.13; lane 13 = S. chartarum CBS 324.65; lane 14 = S. chlorohalonata CBS 413.95; lane 15 = S. chartarum CBS 414.95; lane 16 = no template control. Expected fragment lengths: S. chartarum S type 346 bp, S. chartarum A type 230 bp, S. chlorohalonata 544 bp + 230 bp. Different triplex PCR banding patterns are marked as genotype A (A), genotype S (S), and genotype H (H)
The characteristic amplicons of genotype H were also obtained in single-plex PCRs with the three different primer pairs (Online Resource 4) indicating that the distinct triplex PCR banding pattern of genotype H was not a result of primer interference. DNA was isolated from newly prepared single spore cultures of three S. chartarum genotype H strains as well as from one strain each of the S. chartarum genotypes S and A. The PCR analysis of samples resulted in the expected single amplicons for genotype S and A and confirmed the characteristic double band for genotype H (data not shown). The results confirmed that three separate banding patterns can be detected in strains of S. chartarum and that the double banding pattern observed in genotype H was no artifact from culture contamination. Therefore, the newly developed triplex PCR assay is a useful tool for the differentiation of S. chartarum S, A, and H genotypes.
Genetic analysis of S. chartarum strains
In order to further elucidate the genetic context for the triplex PCR assay, we analyzed the SAT and ATR gene clusters in 13 representative strains of S. chartarum and one strain of S. chlorohalonata. PCR primers were designed for the detection of each of the 21 genes in the SAT gene cluster and of 14 genes in the ATR gene cluster in all analyzed strains. Four strains each of S. chartarum genotypes S and H as well as five strains of genotype A and one strain of S. chlorohalonata were analyzed by PCR. Figure 2 shows a schematic representation of the configuration of SAT- and ATR genes in the different S. chartarum genotypes. Details on the presence (+) or absence (−) of each of the 21 SAT genes are depicted in Online Resource 5. The respective results for the 14 ATR genes are presented in Online Resource 6. Results showed that the four analyzed S. chartarum genotype S strains produced PCR signals for all 21 SAT genes. These strains were negative for all the 14 ATR genes (Online Resource 6). All five analyzed S. chartarum genotype A strains were PCR positive for all ATR genes (Online Resource 6). Three of these strains were negative for all SAT genes. However, strains S1286 and CBS 129.13 showed amplification products of the expected size for some but not all of the SAT genes. Neither of the two strains was PCR positive for any of the genes in the SAT core cluster 2 (SC2, genes SAT11–SAT16, according to Semeiks et al. 2014).

Schematic representation of the configuration of SAT genes and ATR genes in S. chartarum genotypes and in S. chlorohalonata according to PCR analysis. Blue, green, and orange boxes represent satratoxin core gene clusters SC1, SC22, and SC3, respectively. Red boxes represent the atranone core gene cluster. White areas represent missing genes
S. chlorohalonata CBS 413.95 was originally assigned to S. chartarum according to the reference culture collection. However, the strain had the banding pattern typical for the former species in the triplex PCR. The strain was PCR negative for all analyzed SAT genes indicating either that this gene cluster is absent in S. chlorohalonata or that genes are divergent from S. chartarum, so that it cannot be detected by our PCR assay. Regarding the atranone gene cluster, S. chlorohalonata CBS 413.95 was positive for eight of the 14 ATR genes (genes ATR1, 3, 4, 5, 7, 8, 11, 14) but negative for six other ATR genes (ATR2, 6, 9, 10, 12, 13). This finding suggests that some of the ATR genes were either missing in this species or genetically different from their homologs in S. chartarum genotype A and, therefore, do not allow amplification by PCR with the applied primers.
Strains of S. chartarum genotype H showed a distinct pattern of genes: all genes of the SC1 cluster (SAT1–SAT10) were detectable by PCR in the analyzed strains, whereas no PCR products were obtained for all six genes of the SC2 cluster (SAT11–SAT16). For the SC3 cluster (SAT17–SAT21), four of five genes were detectable in all strains. SAT21-specific PCR products were obtained for only two of the four genotype H strains. It was especially striking to see that the genotype H strains completely lacked the genes in the SC2 cluster and that this fact was correlated with their inability to produce macrocyclic trichothecenes (see Table 1). According to the data of this study, these mycotoxins were exclusively produced by S. chartarum genotype S strains, suggesting that the SAT11–SAT16 genes are inevitable for the biosynthesis of macrocyclic trichothecenes. Moreover, they might be a useful genetic marker for high-cytotoxic strains of this species.
The genotype concept as developed during the current study is not fully congruent with the previously existing chemotype concept in S. chartarum because the new genotype H was not reflected as a type of its own. According to Desjardins (2008), independent parallel analysis using PCR-based techniques and chemical analysis of toxin production need to be performed to characterize a new genotype in mycotoxin-producing fungi. In the current study, analysis of the genetic configuration and ability to produce macrocyclic trichothecenes has revealed that the newly described genotype is unique among chemotype A strains of the species. Likewise, it is clearly genetically separate from chemotype A strains that harbor only the genes coding for atranone biosynthesis. During the current study, only a limited number of strains of the new genotype have been analyzed, both with respect to their genetic configuration and production of macrocyclic trichothecenes. It may therefore be possible that isolates exist in the field harboring some of the genes in SC2. Moreover, isolates may exist which can produce and excrete intermediates of the pathway for macrocyclic trichothecenes. Also, strains may exist that have replaced the function of missing genes by orthologues, as has been described in Fusarium spp. (Alexander et al. 2009). However, based on the data presented here, this is highly speculative and should therefore be further elucidated by analysis of a wider collection of isolates.
Data presented here cannot answer the question of the genetic origin of the new genotype. Three possible explanations for the observed genetic configuration in genotype H can be suggested, (i) genotype H has evolved independently from genotypes S and A, (ii) genotype H has evolved from genotype A strains that have picked up SC1 and SC3 by horizontal gene transfer, and (iii) genotype H has evolved from genotype S strains that have lost SC2 and picked up the complete ATR gene cluster by horizontal gene transfer. Such transfer of complete mycotoxin gene clusters was described for other mycotoxigenic fungi (Slot and Rokas 2011).
The existence of the new genotype H in S. chartarum challenges the concept of mutual exclusiveness of chemotype-specific gene clusters in S. chartarum (Semeiks et al. 2014). The results presented here show that also the differentiation of chemotypes, based on a nucleotide polymorphism in the TRI5 gene of S. chartarum, does not fully correlate with the ability of strains to produce highly toxic satratoxins (Andersen et al. 2003). This observation is in agreement with studies by Peltola et al. (Peltolta et al. 2002) who developed a PCR assay based on the TRI5 gene in S. chartarum and found that 40% of the PCR-positive isolates did not produce any macrocyclic trichothecenes. Since the PCR primers applied in this study were specific for type S strains, it can be speculated that the non-producers might have been strains of the new genotype H. Results obtained during the current study have shown that macrocyclic trichothecenes were only produced by strains identified as genotype S using the newly developed triplex PCR assay. It was demonstrated that the identification of strains that is based on sequence analysis of the TRI5 gene does not differentiate properly between strains of genotypes S and H and will therefore overestimate the number of satratoxin producers among strains of S. chartarum.
It can be concluded here that the new triplex PCR assay provides a more reliable method for the identification of highly toxic S. chartarum strains. Moreover, the results obtained will be of great help in the development of DNA-based assays that can be useful as tools for the rapid and specific detection of satratoxin-producing S. chartarum strains in environmental samples.
Funding information
Sponsored by the German Federal Ministry of Education and Research (BMBF) (grant no. 01PL17016) and the Brigitte and Wolfram Gedek Foundation, Ismaning, Germany.
Change history
13 February 2020
The original version of this paper was published with error. The supplementary materials originally provided were not captured during article production.
References
Abbas HK, Johnson BB, Shier WT, Tak H, Jarvis BB, Boyette CD (2002) Phytotoxicity and mammalian cytotoxicity of macrocyclic trichothecene mycotoxins from Myrothecium verrucaria. Phytochem 59:309–313
Aleksic B, Bailly S, Draghi M, Pestka JJ, Oswald IP, Robine E, Bailly JD, Lacroix MZ (2016) Production of four macrocyclic trichothecenes by Stachybotrys chartarum during its development on different building materials as measured by UPLC-MS/MS. Build Environ 106:265–273
Alexander NJ, Proctor RH, McCormick SP (2009) Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxicol Rev 28:198–215
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andersen B, Nielsen KF, Thrane U, Szaro T, Taylor JW, Jarvis BB (2003) Molecular and phenotypic descriptions of Stachybotrys chlorohalonata sp. nov. and two chemotypes of Stachybotrys chartarum found in water-damaged buildings. Mycologia 95:1227–1258
Andersen B, Frisvad JC, Søndergaard I, Rasmussen IS, Larsen LS (2011) Associations between fungal species and water-damaged building materials. Appl Environ Microbiol 77:4180–4188
Biermaier B, Gottschalk C, Schwaiger K, Gareis M (2015) Occurrence of Stachybotrys chartarum chemotype S in dried culinary herbs. Mycotox Res 31:23–32
Bruker Daltoniks (2013) MALDI Biotyper 3.1 user manual, Bruker Daltoniks GmbH
Centers for Disease Control and Prevention (2002) Pulmonary hemorrhage/hemosiderosis among infants Cleveland, Ohio, 1993/1996. Morb Mort Weekly Rep 49:180–184
Cruse M, Telerant R, Gallagher T, Lee T, Taylor JW (2002) Cryptic species in Stachybotrys chartarum. Mycologia 94:814–822
Dearborn DG, Yike I, Sorenson WG, Miller MJ, Etzel RA (1999) Overview of investigations into pulmonary hemorrhage among infants in Cleveland, Ohio. Environ Health Perspect 107:495–499
Dearborn DG, Smith PG, Dahms BB, Allan TM, Sorsenson WG, Montana E, Etzel RA (2002) Clinical profile of 30 infants with acute pulmonary hemorrhage in Cleveland. Pediatrics 110:627–637
Desjardins AE (2008) Natural product chemistry meets genetics: when is a genotype a chemotype? J Agric Food Chem 56:7587–7592
El-Kady IA, Moubasher MH (1982) Toxigenicity and toxins of Stachybotrys isolates from wheat straw samples in Egypt. Exp Mycol 6:25–30
Etzel RA (2003) Stachybotrys. Curr Op Pediat 15:103–106
Etzel RA, Montana E, Sorenson WG, Kullman GJ, Allan TM, Dearborn DG, Olson DR, Jarvis BB, Miller JD (1998) Acute pulmonary hemorrhage in infants associated with exposure to Stachybotrys atra and other fungi. Arch Pediatr Adolesc Med 152:757–762
Forgacs J, Carll WT, Herring AS, Hinshaw WR (1958) Toxicity of Stachybotrys atra for animals. Trans New York Acad Sci 20:787–808
Gareis M, Gottschalk C (2014) Stachybotrys spp. and the guttation phenomenon. Mycotox Res 30:151–159
Gravesen S, Nielsen PA, Iversen R, Nielsen KF (1999) Microfungal contamination of damp buildings - examples of risk constructions and risk materials. Environ Health Perspect 107:505–508
Gruenwald M, Rabstein A, Remesch M, Kuever J (2015) MALDI-TOF mass spectrometry fingerprinting: a diagnostic tool to differentiate dematiaceous fungi Stachybotrys chartarum and Stachybotrys chlorohalonata. J Microbiol Methods 115:83–88
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hanelt M, Gareis M, Kollarczik B (1994) Cytotoxicity of mycotoxins evaluated by the MTT-cell culture assay. Mycopathologia 128:167–174
Hernandez F, Cannon M (1982) Inhibition of protein synthesis in Saccharomyces cerevisiae by the 12,13-epoxytrichothecenes trichodermol, diacetoxyscirpenol and verrucarin A. Reversibility of the effects. J Antibiot 35:875–881
Hinkley SF, Mazzola EP, Fettinger JC, Lam Y-F, Jarvis BB (2000) Atranones A–G, from the toxigenic mold Stachybotrys chartarum. Phytochemistry 55:663–673
Hinkley SF, Moore JA, Squillari J, Tak H, Oleszewski R, Mazzola EP, Jarvis BB (2003) New atranones from the fungus Stachybotrys chartarum. Magn Reson Chem 41:337–343
Hintikka E-L (1977a) The genus Stachybotrys. In: Wyllie TD, Morehouse LG (eds.) Mycotoxic Fungi, mycotoxins, Mycotoxicosis. An Encyclopedia Handbook. . New York, Basel, Marcel Dekker Inc., 91–98
Hintikka E-L (1977b) Stachybotryotoxicosis in horses. In: Wyllie TD, Morehouse LG (eds) Mycotoxic fungi, mycotoxins, mycotoxicosis. An encyclopedia handbook. Marcel Dekker Inc., New York, Basel, pp 181–185
Jarvis BB, Salemme J, Morais A (1995) Stachybotrys toxins. 1. Nat Toxins 3:10–16
Johanning E, Yang CS (1994) Health problems related to fungal exposure - the example of toxigenic Stachybotrys chartarum. Eastern New York Occup health Prog—CHP, 169-182
Johanning E, Landsbergis P, Gareis M, Yang CS, Olmsted E (1999) Clinical experience and results of a sentinel health investigation related to indoor fungal exposure. Environ Health Perspect 107(Suppl 3):489–494
Kriek NPJ, Marasas WFO (1983) Field outbreak of ovine stachybotryotoxicosis in South Africa. In: Ueno Y (ed) Thrichothecenes - chemical, biological and toxicological aspects. Elsevier Science Ltd, pp 279–284
Lombard L, Houbraken J, Decock C, Samson RA, Meijer M, Reblova M, Groenewald JZ, Crous PW (2016) Generic hyper-diversity in Stachybotriaceae. Persoonia 36:156–246
Miller JD, Rand TG, Jarvis BB (2003) Stachybotrys chartarum: cause of human disease or media darling? Med Mycol 41:271–291
Nielsen KF, Holm G, Uttrup LP, Nielsen PA (2004) Mould growth on building materials under low water activities. Influence of humidity and temperature on fungal growth and secondary metabolism. Int Biodeterior Biodegrad 54:325–336
Niessen L, Vogel RF (2010) Detection of Fusarium graminearum DNA using a loop-mediated isothermal amplification (LAMP) assay. Int J Food Microbiol 140:183–191
Nikulin M, Pasanen A-L, Berg S, Hintikka E-L (1994) Stachybotrys atra growth and toxin production in some building materials and fodder under different relative humidities. Appl Environ Microbiol 60:3421–3424
Peltolta J, Niessen L, Nielsen KF, Jarvis BB, Andersen B, Salkinoja-Salonen M, Möller EM (2002) Toxigenic diversity of two different RAPD groups of Stachybotrys chartarum isolates analyzed for potential for trichothecene production and for boar sperm cell motility inhibition. Can J Microbiol 48:1017–1029
Rand TG, Flemming J, David Miller J, Womiloju TO (2006) Comparison of inflammatory responses in mouse lungs exposed to atranones A and C from Stachybotrys chartarum. J Toxicol Environ Health Part A 69:1239–1251
Robert V, Stegehuis G, Stalpers J (2005) The MycoBank engine and related databases. http://www.mycobank.org. Accessed 13.02.2019
Rocha O, Ansari K, Doohan FM (2005) Effects of trichothecene mycotoxins on eukaryotic cells: a review. Food Addit Contam 22:369–378
Schneider DJ, Marasas WFO, Kuys JCD, Kriek NPJ, Van Schalkwyk GC (1979) A field outbreak of suspected stachbotryotoxicosis in sheep. J South Af Vet Assoc 50:73–81
Semeiks J, Borek D, Otwinowski Z, Grishin NV (2014) Comparative genome sequencing reveals chemotype-specific gene clusters in the toxigenic black mold Stachybotrys. BMC Genomics 15:590–606
Slot JC, Rokas A (2011) Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr Biol 21:134–139
Ueno Y (1977) Mode of action of trichothecenes. Ann Nutr Aliment 31:885–900
Ulrich S, Biermaier B, Bader O, Wolf G, Straubinger RK, Didier A, Sperner B, Schwaiger K, Gareis M, Gottschalk C (2016) Identification of Stachybotrys spp. by MALDI-TOF mass spectrometry. Anal Bioanal Chem 408:7565–7581
Vertinsky KI (1940) Food poisoning in horses due to Stachybotrys alternans. Sovet Vet 5:61–68
Vesper SJ, Magnuson ML, Dearborn DG, Yike I, Hauglan RA (2001) Initial characterization of the hemolysin stachylysin from Stachybotrys chartarum. Infect Immun 69:912–916
Wang Y, Hyde KD, McKenzie EH, Jiang Y-L, Li D-W, Zhao D-G (2015) Overview of Stachybotrys (Memnoniella) and current species status. Fungal Divers 71:17–83
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform 13:134–145
Acknowledgments
We wish to thank Prof. Dr. Frank Ebel and Prof. Dr. Reinhard Straubinger, Ph.D., for the critical reading of the manuscript and Mrs. Elisabeth Hamzi-Schmidt for the proofreading and corrections.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ulrich, S., Niessen, L., Ekruth, J. et al. Truncated satratoxin gene clusters in selected isolates of the atranone chemotype of Stachybotrys chartarum (Ehrenb.) S. Hughes. Mycotoxin Res 36, 83–91 (2020). https://doi.org/10.1007/s12550-019-00371-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12550-019-00371-x