
Abstract
Expression of Bcl-xL correlates with the clinical outcomes of patients with cancer. While the role of Bcl-2 in angiogenesis is becoming increasingly evident, the function of Bcl-xL in angiogenesis is unclear. Here, we showed that epidermal growth factor (EGF) induces in vitro capillary sprouting and Bcl-xL expression in primary endothelial cells. Bcl-xL-transduced human dermal microvascular endothelial cells (HDMEC-Bcl-xL), but not empty vector control cells, spontaneously organize into capillary-like sprouts. Searching for a mechanism to explain these responses, we observed that Bcl-xL induced expression of the pro-angiogenic chemokines CXC ligand-1 (CXCL1) and CXC ligand-8 (CXCL8), and that blockade of CXC receptor-2 (CXCR2) signaling inhibited spontaneous sprouting of HDMEC-Bcl-xL. Bcl-xL led to Bcl-2 upregulation, but Bcl-2 did not upregulate Bcl-xL, suggesting the existence of a unidirectional crosstalk from Bcl-xL to Bcl-2. EGF and Bcl-xL activate the mitogen-activated protein kinase/ERK pathway resulting in upregulation of vascular endothelial growth factor (VEGF), a known inducer of Bcl-2 in endothelial cells. Inhibition of VEGF receptor signaling in HDMEC-Bcl-xL prevented Bcl-2 upregulation and demonstrated the function of a VEGF-mediated autocrine loop. Bcl-2 downregulation by RNAi blocked CXCL1 and CXCL8 expression downstream of Bcl-xL, and markedly decreased angiogenesis in vivo. We conclude that Bcl-xL functions as a pro-angiogenic signaling molecule controlling Bcl-2 and VEGF expression. These results emphasize a complex interplay between Bcl-2 family members beyond their classical roles in apoptosis.
Similar content being viewed by others


Main
Deregulated angiogenesis is a major factor in the pathobiology of cancer. Tumor cells use several strategies to acquire and sustain their own blood vessel network that provides the means for tumor progression and metastatic spread. One of these strategies is to enhance the survival of endothelial cells.1, 2 Indeed, it is common to find areas in solid tumors in which the only nontumor cells that are capable of surviving to the constraints of the tumor microenvironment are the cells that compose blood vessels. Vascular endothelial growth factor (VEGF) plays a predominant role in the regulation of endothelial cell survival.2, 3, 4, 5 Many tumors present exquisite dependency on VEGF signaling. An elegant study6 demonstrated that tumor-associated endothelial cells began to undergo apoptosis before the cancer cells, demonstrating a role for VEGF in endothelial cell survival. Such findings underscore the relevance of VEGF to endothelial cell survival and tumor biology.
Signaling pathways involved in VEGF's pro-survival effects on endothelial cells are becoming better understood. VEGF binding to VEGFR2 activates a PI3K/Akt mediated signaling pathway that results in the upregulation of the pro-survival Bcl-2 protein.4, 5 Bcl-2 protects endothelial cells against death induced by removal of growth factors,5 and against thrombospondin-1 induced apoptosis.7 Furthermore, therapeutic inhibition of Bcl-2 function with a small molecule inhibitor (i.e. TW37) is sufficient to induce endothelial cell apoptosis.8 Notably, constitutive upregulation of Bcl-2 expression in vascular endothelial cells enhances tumor angiogenesis and accelerate tumor progression.9
We recently demonstrated that the effect of Bcl-2 on angiogenesis is not limited to its antiapoptotic role. Bcl-2 activates nuclear factor-κB (NF-κB) transcriptional activity, which leads to upregulation of expression of the pro-angiogenic chemokines CXC ligand-1 (CXCL1) and CXC ligand-8 (CXCL8).10 These chemokines function in an autocrine signaling pathway mediated by CXC receptor-2 (CXCR2) that enhances the angiogenic potential of endothelial cells. Interestingly, sub-apoptotic concentrations of a small molecule inhibitor of Bcl-2 (i.e. TW37) inhibit CXCL1 and CXCL8 upregulation, and partially block capillary sprouting induced by VEGF in vitro.8 Taken together, these data demonstrate the role of Bcl-2 as a pro-angiogenic signaling molecule.
Bcl-xL and Bcl-2 share high sequence homology.11 Furthermore, Bcl-xL and Bcl-2 function as pro-survival factors via a common pathway.12 Comparable phenotypes were observed in transgenic mice overexpressing either Bcl-2 or Bcl-xL.13 However, Bcl-2 and Bcl-xL are differentially regulated. VEGF induces Bcl-2 expression in endothelial cells, but it does not affect Bcl-xL expression.5 In contrast, epidermal growth factor (EGF) induces expression of Bcl-xL in many cell types, including parthenotes and keratinocytes.14, 15
Bcl-2 and Bcl-xL were initially described to be interchangeable in their abilities to block apoptosis. However, recent studies suggest that their binding partners and roles in cell survival may be cell-type and stimuli-dependent. With respect to angiogenesis, the role of Bcl-xL is unclear. Here, we demonstrate that EGF induces Bcl-xL expression in endothelial cells. Subsequently, we show that Bcl-xL induces VEGF expression through the mitogen-activated protein kinase (MAPK) signaling pathway. VEGF binds to VEGFR2, and induces expression of Bcl-2 and the angiogenic chemokines CXCL1 and CXCL8. These two chemokines, together with VEGF, activate autocrine-signaling pathways that enhance the angiogenic potential of endothelial cells in vitro and in vivo. Taken together, these data demonstrate that Bcl-xL mediates a signaling pathway that is upstream from Bcl-2 and contributes to angiogenesis through a process that is functionally independent from the role of these molecules on endothelial cell survival.
Results
Bcl-xL induces capillary sprouting in vitro
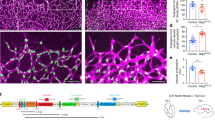
Before beginning studies on the function of Bcl-xL in angiogenesis, we searched for physiological inducers of this protein in endothelial cells. We observed that EGF induces Bcl-xL expression in primary endothelial cells (Figure 1a). To evaluate if Bcl-xL enhances the angiogenic potential of endothelial cells, we performed in vitro capillary sprouting assays. For these assays, we used several pools of primary human dermal microvascular endothelial cells (HDMEC) stably transduced with Bcl-xL (HDMEC-Bcl-xL), or empty-vector control cells (HDMEC-LXSN) (Figure 1b and j). We observed that HDMEC-Bcl-xL cells spontaneously developed capillary-like sprouts in vitro (Figure 1b and f). In contrast, empty vector control cells (HDMEC-LXSN) did not sprout in collagen (Figure 1b and d). The positive controls for this experiment were HDMEC-Bcl-2, cells with strong angiogenic potential.10 Notably, the increase in capillary sprouting cannot be simply attributed to an increase in cell number as measured by the sulforhodamine B (SRB) assay (Figure 1c). EGF is a strong inducer of endothelial cell capillary sprouting in vitro (Figure 1g). Notably, the angiogenic potential of EGF was significantly inhibited in primary endothelial cells transduced with short hairpin RNA for Bcl-xL (Figures 1g and 6a). Taken together, these data demonstrate that Bcl-xL acts as a pro-angiogenic molecule, and that Bcl-xL is involved in EGF-induced angiogenesis.

Bcl-xL induces capillary sprouting in vitro. (a) Expression of Bcl-xL analyzed by immunoblotting of lysates obtained from HDMEC-Bcl-xL, HDMEC-LXSN (empty vector control) and HDMEC-LXSN exposed to 50 ng/ml EGF. (b) Capillary sprouting assay with HDMEC-Bcl-2, HDMEC-Bcl-xL and HDMEC-LXSN plated on type I collagen. At daily intervals, the number of sprouts was counted in six random microscopic fields (× 100) from triplicate wells/condition. The data presented are representative of three independent experiments. (c) SRB assays were performed with HDMEC-Bcl-2, HDMEC-Bcl-xL, or HDMEC-LXSN to evaluate relative cell number per condition. (d–f) Representative microscopic fields (× 200) of untreated HDMEC-LXSN (d), HDMEC-Bcl-2 (e), or HDMEC-Bcl-xL (f). Arrows point to capillary sprouts. (g) Capillary sprouting assay with HDMEC stably transduced with shRNA-Bcl-xL or scrambled control (shRNA-C) and cultured in the presence of 0, 50, or 100 ng/ml EGF. As controls, HDMEC transduced with shRNA-C were cultured in presence of 50 ng/ml VEGF. (h) Capillary sprouting assay with HDMEC stably transduced with wild-type Bcl-xL, or with Bcl-xL mutants L130A and XB. HDMEC stably transduced with the empty retroviral vector (LXSN) were used as negative controls. (i) Western blot analysis of two independent pools of HDMEC-Bcl-xL (L130A) and HDMEC-Bcl-xL (XB) cells using either anti-Bcl-xL or anti-HA antibody. (j) Western blot analysis of three independent pools of HDMEC-Bcl-xL and HDMEC-Bcl-2 cells. (k) Expression of Bcl-2 and Bcl-xL analyzed by immunoblotting of lysates from HDMEC transiently transfected with Bcl-xL

Bcl-xL enhances angiogenesis in vivo. The SCID mouse model of human angiogenesis was used to evaluate the effects of Bcl-xL on angiogenesis in vivo. (a) Western blot to evaluate the effect of lentiviruses encoding shRNA-Bcl-xL, or control shRNA-C, on Bcl-xL expression levels in primary endothelial cells (HDMEC). (b) Quantification of microvessel density of the scaffolds immunostained with anti-factor VIII antibody. Statistical significance was determined at P⩽0.05. (c and d) Immunohistochemistry for factor VIII (red stain) to identify blood vessels in scaffolds seeded with HDMEC transduced with scrambled sequence control shRNA-C (c) or with shRNA-Bcl-xL. (d) Arrowheads point to factor VIII-positive blood vessels. Photomicrographs were taken at × 200
To begin to understand mechanisms underlying the angiogenic activity of Bcl-xL, we stably transduced primary endothelial cells with the Bcl-xL mutants L130A or XB.16 L130A mutation disrupts a residue that is directly involved in forming hydrophobic interactions with the BH3 domain of death agonists, such as Bax. Despite losing its ability to dimerize with Bax, this mutant was shown to maintain some level of protection against apoptosis.16 The XB mutation was generated by replacing the region surrounding the Bcl-xL helix 5 and helix 6 hairpin with the corresponding region from Bax. In contrast to the L130A mutant, XB retained its ability to dimerize with Bax and present an antiapoptotic effect similar to wild-type Bcl-xL.16 Here, we observed that endothelial cells stably transduced with either L130A or XB (Figure 1i) showed a similar decrease in their in vitro angiogenic potential, suggesting that the effect of Bcl-xL in angiogenesis is not dependent on its ability to dimerize with the BH3 domain of death agonists (Figure 1h).
Unexpectedly, we found that Bcl-2 is upregulated in HDMEC-Bcl-xL cells (Figure 1j) while performing Western blots for the characterization of the pools of stably transduced endothelial cells. In contrast, Bcl-xL was not upregulated in HDMEC-Bcl-2 cells (Figure 1j). To confirm these results, and eliminate the possibility that the upregulation of Bcl-2 in HDMEC-Bcl-xL cells was an artifact caused by the retroviral infection of these cells, we performed a time-course experiment in which we transiently transfected Bcl-xL in HDMEC (Figure 1k). We observed that the increase in Bcl-xL expression preceded the upregulation of Bcl-2 (Figure 1k). Taken together, these findings suggested a potential explanation for the spontaneous capillary sprouting observed when HDMEC-Bcl-xL cells were cultured in collagen. We know that Bcl-2 induces CXCL1 and CXCL8 expression in endothelial cells, and that these chemokines work through an autocrine pro-angiogenic signaling pathway mediated by CXCR2.10 Therefore, if Bcl-xL induces Bcl-2 expression, it could potentially activate this angiogenic loop, explaining the spontaneous sprouting phenotype that was observed with HDMEC-Bcl-xL.
Bcl-xL activates a pro-angiogenic signaling pathway mediated by CXCL1 and CXCL8 and binding to CXCR2
To test the hypothesis that Bcl-xL activates angiogenesis through a chemokine-mediated signaling pathway, we evaluated expression of CXCL1 and CXCL8 in endothelial cells stably transduced with Bcl-xL. We observed that indeed CXCL1 and CXCL8 were upregulated at the mRNA (Figure 2a and c) and at the protein levels (Figure 2b and d) in HDMEC-Bcl-xL cells, as compared to controls. It is known that CXCR2 is a receptor that mediates CXCL1 and CXCL8 pro-angiogenic signaling.17, 18 To verify whether Bcl-xL-induced CXCL1 and CXCL8 promote angiogenesis through an autocrine pathway mediated by CXCR2, we allowed HDMEC-Bcl-xL cells to sprout in collagen for 4 days, and then blocked this receptor using a neutralizing antibody. We observed CXCR2 blockade caused a marked decrease in the number of capillary sprouts, as compared to IgG-treated control cells (Figure 2e). Anti-CXCR2 antibody treatment did not affect endothelial cell viability, demonstrating that the disruption of sprouts observed above was not due to cell death (Figure 2f). These results suggest that Bcl-xL increases the angiogenic potential of endothelial cells via upregulation of CXC pro-angiogenic chemokines. The next question that we attempted to answer was how Bcl-xL induces Bcl-2 expression in endothelial cells.

Bcl-xL activates a pro-angiogenic signaling pathway mediated by CXCL1 and CXCL8 and binding to CXCR2. (a and c) mRNA relative expression of CXCL1 (a) and CXCL8 (c) in HDMEC-Bcl-xL and HDMEC-LXSN, as determined by Affymetrix microarray. (b and d) ELISA for evaluation of CXCL1 (b) and CXCL8 (d) expression in HDMEC-Bcl-xL and HDMEC-LXSN. (e) Capillary sprouting assays performed with HDMEC-LXSN and HDMEC-Bcl-xL. Starting on the 4th day, and continuing thereafter, cells were exposed to either 1 μg/ml anti-CXCR2 or 1 μg/ml non-specific IgG. At daily intervals, the number of sprouts was counted in six random fields from triplicate wells/condition. (f) SRB assays for evaluation of viability of HDMEC-LXSN, HDMEC-Bcl-2 and HDMEC-Bcl-xL after exposure to either 1 μg/ml g anti-CXCR2, or 1 μg/ml IgG for 48 h. Data reflect analysis of triplicate wells/condition, and is representative of three independent experiments
Bcl-xL-induced upregulation of Bcl-2 is mediated by VEGF
VEGF induces Bcl-2 expression in endothelial cells.5 Here, we hypothesized that Bcl-xL utilizes this pathway to induce Bcl-2 expression. We transiently transfected primary endothelial cells with pcDNA3-Bcl-xL, or with an empty vector control, and evaluated VEGF expression. We observed a significant increase in VEGF expression at all time periods evaluated (9–24 h) in Bcl-xL transfected cells, as compared to empty vector control (Figure 3a). To evaluate specificity of this response, we infected HDMEC-Bcl-xL cells with lentiviruses expressing a short hairpin interfering RNA (shRNA) against Bcl-xL (Figure 3b), and observed that VEGF upregulation was completely inhibited. We repeated this experiment with endothelial cells exposed to EGF, and observed that this physiological stimulus results in upregulated VEGF expression. When EGF-induced Bcl-xL expression was prevented with shRNA-Bcl-xL (Figure 3b), EGF lost its ability to induce VEGF upregulation (Figure 3c).

Bcl-xL-induced upregulation of Bcl-2 is mediated by VEGF expression. (a) ELISA for evaluation of VEGF expression in endothelial cells 9–24 h after transient transfection with Bcl-xL. (b) Western blot of HDMEC-Bcl-xL and HDMEC-LXSN exposed to 50 ng/ml EGF after exposure to lentiviruses encoding shRNA-Bcl-xL or control vectors (shRNA-C). (c) ELISA to evaluate VEGF expression levels in HDMEC-Bcl-xL, HDMEC-LXSN, or HDMEC-LXSN cultured in presence of 50 ng/ml EGF and exposed to lentiviruses encoding shRNA-Bcl-xL or controls. (d) Western blot to evaluate the effect of blockade of VEGFR2 with increasing concentrations of PTK787/ZK222584 (PTK/ZK) on Bcl-2 expression levels in HDMEC-Bcl-xL, HDMEC-LXSN exposed to 50 ng/ml VEGF or HDMEC-LXSN
To examine the involvement of VEGF on Bcl-xL-induced Bcl-2 upregulation in endothelial cells, we used a small molecule inhibitor of the VEGF tyrosine kinase receptors (PTK787/ZK222584).19 We observed that concentrations of 100 nM PTK787/ZK222584 or higher downregulated Bcl-2 expression in HDMEC-Bcl-xL (Figure 3d). As controls, we exposed endothelial cells to VEGF and observed that 100 nM PTK787/ZK222584 inhibited VEGF-induced Bcl-2 upregulation (Figure 3d). Taken together, these results demonstrate that Bcl-xL induces VEGF expression in endothelial cells, and that an autocrine pathway mediated through VEGFR2 leads to upregulation of Bcl-2 expression levels in these cells.
Bcl-xL induces VEGF upregulation via the MAPK pathway
We next attempted to understand how Bcl-xL induces VEGF expression. Bcl-2 activates the NF-κB signaling pathway,20, 21 and induces CXC chemokine expression in endothelial cells.10 Since Bcl-2 and Bcl-xL are homologous, and the VEGF promoter has binding sites for NF-κB,22 we hypothesized the Bcl-xL-induced VEGF expression was also mediated NF-κB activation. Surprisingly, Bcl-xL did not induce NF-κB activation (data not shown), and inhibition of inhibitor-κB (I-κB) kinase did not affect VEGF expression in HDMEC transiently transfected with Bcl-xL (Figure 4a). In attempt to find an alternative mechanism to explain how Bcl-xL induced VEGF expression, we investigated other signaling cascades with known functions in angiogenesis. By immunoblotting, we found that the levels of phosphorylated p44/p42-α MAPK were increased in HDMEC-Bcl-xL cells (Figure 4b). The induction of the MAPK pathway in Bcl-xL expressing endothelial cells was comparable to the effect of EGF (Figure 4b), suggesting that the phosphorylation levels mediated by Bcl-xL overexpression can also be achieved by a physiological stimulus. Notably, blocking MEK/ERK signaling with the inhibitor PD98059 significantly blocked Bcl-xL-driven activation of VEGF (Figure 4c). These results demonstrate that the MAPK, but not NF-κB, signaling pathway mediates Bcl-xL-induced VEGF expression in endothelial cells.

Bcl-xL induces VEGF upregulation via the MAPK pathway. (a) ELISA for VEGF expression in HDMEC-LXSN transiently transfected with Bcl-xL (6–24 h post-transfection) after treatment with 0 or 0.5 μM I-κB kinase inhibitor peptide, or control peptide. Data were normalized by cell number and results are representative of three independent experiments. (b) Western blot to evaluate MAPK phosphorylation in either HDMEC-LXSN exposed to 0 or 50 ng/ml EGF, or HDMEC-Bcl-xL. (c) ELISA to evaluate VEGF expression in HDMEC-LXSN exposed to 0 or 50 ng/ml EGF or HDMEC-Bcl-xL after inhibition of MEK signaling with 0 or 10 μ M PD98059 (MEK inhibitor)
Downregulation of Bcl-2 expression inhibited upregulation of CXCL1 and CXCL8 in Bcl-xL stably transduced endothelial cells
To evaluate whether Bcl-2 mediates the increase in CXCL1 and CXCL8 expression in Bcl-xL transduced endothelial cells, the endogenous expression of Bcl-2 was blocked with a lentiviral-driven shRNA. Western blot analysis demonstrated that the shRNA-Bcl-2 mediated effective and specific downregulation of Bcl-2 expression, while not affecting Bcl-xL levels (Figure 5a). We observed that Bcl-2 downregulation correlates with a decrease in CXCL1 and CXCL8 expression levels (Figure 5b and c). Indeed, expression of these chemokines in HDMEC-Bcl-xL cells returned to baseline levels when Bcl-2 was downregulated. These data demonstrate that Bcl-xL does not induce CXC chemokines directly, and that Bcl-2 is a required mediator of this pathway.

Downregulation of Bcl-2 expression inhibited upregulation of CXCL1 and CXCL8 in Bcl-xL stably transduced endothelial cells. (a) Western blot to evaluate the effect of lentiviruses encoding shRNA-Bcl-2, or control shRNA-C, on Bcl-2 expression levels in HDMEC-LXSN, HDMEC-Bcl-xL and HDMEC-Bcl-2. (b and c) ELISA to evaluate the expression levels of CXCL1 (b) and CXCL8 (c) in the cells described in panel (a). Data were normalized by cell number and results are representative of three independent experiments
Bcl-xL induces angiogenesis in vivo in a process dependent on Bcl-2 expression
To examine the effect of Bcl-xL on angiogenesis in vivo, we used the severe combined immunodeficient (SCID) mouse model of human angiogenesis.23 This tissue engineering-based model allows for the differentiation of human endothelial cells into functional human blood vessels that anastomize with the mouse vasculature.23 Highly porous biodegradable scaffolds seeded with primary HDMEC transduced with shRNA-Bcl-xL or control lentiviruses (shRNA-C); HDMEC-Bcl-xL transduced with shRNA-Bcl-2 or control lentiviruses; HDMEC-Bcl-2 (positive control)5 or HDMEC transduced with empty vector control LXSN5 were implanted in SCID mice. We observed a significant decrease in microvessel density when Bcl-xL was downregulated in primary endothelial cells (Figure 6a–d). Notably, a significant decrease in vascularization was observed in implants seeded with HDMEC-Bcl-xL transduced with shRNA-Bcl-2, as compared to implants containing HDMEC-Bcl-xL transduced with control lentiviruses (Figure 7a–d). As expected, positive control implants seeded with HDMEC-Bcl-2 were highly vascularized (Figure 7b and e), and implants seeded with control HDMEC-LXSN cells presented low microvascular density (Figure 7b and f). We ran parallel studies and observed that these results cannot be attributed to a decrease in cell viability mediated by infection with shRNA-Bcl-2 or shRNA-C lentiviruses (Figure 7g). Taken together, these data demonstrate that Bcl-xL induces angiogenesis in vivo, and that the effect of Bcl-xL on angiogenesis is mediated through Bcl-2.

Bcl-xL-induced in vivo angiogenesis is dependent on Bcl-2 expression levels. The SCID mouse model of human angiogenesis was used to evaluate the effects of Bcl-xL on angiogenesis in vivo. (a) Macroscopic view of scaffolds seeded with HDMEC-Bcl-xL transduced with shRNA-Bcl-2 or scrambled sequence control lentiviruses (shRNA-C). (b) Quantification of microvessel density using immunostaining with anti-factor VIII antibody to identify vascular endothelial cells. Statistical significance was determined at P⩽0.05. (c–f) Immunohistochemistry for factor VIII (red stain) to identify blood vessels in scaffolds seeded with HDMEC-Bcl-xL transduced with shRNA-Bcl-2 (c), scrambled sequence control shRNA-C (d), control scaffolds seeded with HDMEC-Bcl-2 (e), or HDMEC-LXSN (f). Arrowheads point to factor VIII-positive blood vessels. Photomicrographs were taken at × 400. (g) SRB assay for determination of viability of HDMEC-Bcl-2, HDMEC-LXSN and HDMEC-Bcl-xL transduced with shRNA-Bcl-2 or control lentiviruses (shRNA-C). Results are representative of three independent experiments
Discussion
The role of Bcl-xL as a pro-survival protein has been extensively studied for the last 10 years. Recent evidence suggests that its function might not be limited to regulation of cell death. Here, we demonstrate that Bcl-xL acts as a pro-angiogenic signaling molecule through a complex pathway that is illustrated in Figure 8. Bcl-xL activates the MAPK/ERK signaling pathway resulting in VEGF upregulation. VEGF secreted by the endothelial cells signals through an autocrine signaling pathway mediated by VEGFR2, which results in expression of Bcl-2. Upregulation of Bcl-2 leads to expression of the pro-angiogenic CXCL1 and CXCL8 in endothelial cells.10 CXCL1 and CXCL8 are pro-angiogenic chemokines that signal through the CXCR2 receptor and increase the angiogenic potential of endothelial cells.10, 24 Collectively, these data demonstrate a novel function for Bcl-xL as a mediator of angiogenesis.

Model depicting proposed pro-angiogenic signaling pathway mediated by Bcl-xL. EGF induces Bcl-xL expression in endothelial cells, which in turn result in VEGF upregulation via activation of a MAPK-mediated signaling pathway. VEGF is secreted, binds to VEGFR2, and induces Bcl-2 expression via an autocrine loop. Bcl-2 upregulation leads to activation of NF-κB pathway and expression of the pro-angiogenic chemokines CXCL1 and CXCL8. The proposed model suggests that both Bcl-xL and Bcl-2 are potential therapeutic targets for anti-angiogenic strategies
Tumor endothelial cells express EGFR and are responsive to pro-angiogenic stimuli mediated by EGF.25 Here, we report that EGF induces Bcl-xL expression in primary endothelial cells, and has a potent angiogenic effect in vitro. Endothelial cells transduced with shRNA-Bcl-xL were significantly less responsive to the pro-angiogenic signal mediated by EGF than endothelial cells transduced with an empty vector control lentivirus. However, the shRNA-Bcl-xL did not block completely the pro-angiogenic effect of EGF. We speculate that the observed lack of complete inhibition might be due to the fact that EGF engages a parallel Bcl-xL-independent angiogenic pathway, or due to lack of complete blockade of Bcl-xL expression with the RNAi.
Bcl-xL stably transduced endothelial cells sprout spontaneously in capillary-like assays. Several pieces of evidence suggest that these results cannot be simply attributed to the well-known role of Bcl-xL as a pro-survival protein: (a) we observed a nine-fold increase in the number of capillary sprouts formed by HDMEC-Bcl-xL, as compared to empty vector control cells. The increase in the number of sprouts happened in absence of any increase in number of cells, as demonstrated by SRB assays. These data demonstrate that Bcl-xL's pro-angiogenic effect does not depend on reducing endothelial cell turnover and therefore increasing the number of cells available for making capillary-like sprouts. (b) We evaluated here two Bcl-xL mutants for their ability to induce angiogenesis in vitro. While the L130A mutation impedes Bcl-xL to dimerize with the BH3 domain of proapoptotic proteins (e.g. Bax), the XB mutation does not prevent this interaction.16 The L130A mutant was shown to retain some protective effect against IL-3 deprivation in IL-3-dependent cells. But, the L130A mutant was not as protective as the XB mutant, which prevented apoptosis of the IL-3-dependent cells as effectively as wild-type Bcl-xL.16 Here, we observed that endothelial cells stably transduced with either L130A or XB showed a similar decrease in their in vitro angiogenic potential, as compared to cells transduced with wild-type Bcl-xL. These data demonstrate that the effect of Bcl-xL in angiogenesis does not depend on its ability to dimerize with the BH3 domain of proapoptotic proteins, which is one of the primary mechanisms by which Bcl-xL protects cells against apoptosis. (c) Sprouting of endothelial cells in collagen requires that the cells align themselves, and organize into tubular structures. This involves cytoskeleton rearrangement and migration of endothelial cells. Recent publications have demonstrated a potential interaction between the Bcl-2-family of proteins and the cytoskeleton.26 We have recently reported that a small molecule inhibitor of Bcl-2 (TW-37) inhibits endothelial cell migration.8 Notably, TW37 inhibited endothelial cell migration at very low, sub-apoptotic, concentrations.8 These data suggest that Bcl-2-mediated inhibition of migration is a process functionally independent of its role as a pro-survival protein. Since Bcl-xL's pro-angiogenic effect is mediated by Bcl-2, these data suggest that Bcl-xL induces angiogenesis through a process that is not dependent on its antiapoptotic activity.
Our experiments demonstrated the existence of a crosstalk between Bcl-xL and Bcl-2 in endothelial cells. Notably, this crosstalk is unidirectional since Bcl-xL induces Bcl-2, but not vice versa. The fact that the crosstalk is mediated by VEGF explained the unidirectionality of the pathway, since we know that VEGF induces Bcl-2, but not Bcl-xL, in endothelial cells.5 It also suggests that this pathway might be unique to endothelial cells. VEGFR2, the receptor that mediates Bcl-2 upregulation in response to VEGF stimulation,4 is fairly specific to endothelial cells.27, 28 This intriguing finding is puzzling from a conceptual standpoint, since Bcl-2 and Bcl-xL are two proteins with similar functions in cell survival, and might be considered redundant. We hypothesize that endothelial cells benefit from a mechanism of crosstalk between these two proteins to exploit their role on angiogenesis. Bcl-xL induces VEGF expression, and Bcl-2 induces CXCL1 and CXCL8. VEGF and these two chemokines are among the most potent angiogenic factors known to date, and together these factors mediate a strong signal for endothelial cells to proliferate, differentiate and organize themselves into neovessels.
We observed that HDMEC-Bcl-xL showed enhanced capillary sprouting in vitro and angiogenesis in vivo than control HDMEC-LXSN cells, but not to the same extent as HDMEC-Bcl-2 cells. These data correlate well with the observation that HDMEC-Bcl-2 cells show higher expression of CXCL1 and CXCL8 than HDMEC-Bcl-xL. These findings were somewhat unexpected, since we observed that Bcl-2 was upregulated in endothelial cells expressing Bcl-xL. We speculate that these differences might be due to the fact that Bcl-xL does not induce as much Bcl-2 as the level of expression of this protein observed in HDMEC-Bcl-2 cells.
The mechanisms involved in VEGF upregulation mediated by Bcl-xL are not completely understood. Some studies have suggested a role for either the ERK, or the NF-κB pathway, in the regulation of VEGF and Bcl-xL expression.29, 30, 31, 32 Here, we did not observe an increase in NF-κB activity in Bcl-xL transiently transfected cells, and did not observe a significant decrease in VEGF expression after blockade of the NF-κB pathway. In contrast, our results demonstrate that MAPK signaling was activated in HDMEC-Bcl-xL and in endothelial cells exposed to EGF, and that blockade of MAPK signaling downregulates VEGF expression in these cells. Notably, while PD98059 abolished EGF-induced VEGF expression, this MAPK inhibitor was only partially effective in preventing VEGF upregulation in HDMEC-Bcl-xL. We speculate that this difference in response might be due to the fact that constitutive overexpression of Bcl-xL engages parallel signaling pathways in addition to the MAPK that also result in VEGF upregulation and that were not inhibited by PD98059. Our data suggest that there is a significant difference in signaling pathways initiated by Bcl-xL as compared to pathways mediated by Bcl-2. While Bcl-2 induces NF-κB activity,10, 20, 21 Bcl-xL signals primarily through the MAPK pathway in endothelial cells. These findings suggest that Bcl-xL and Bcl-2 are not redundant from an angiogenic signaling standpoint. Rather, they complement each other.
Primary endothelial cells transduced with shRNA-Bcl-xL were less angiogenic in vivo, as compared to cells transduced with an empty vector control lentivirus. As expected, when we overexpressed Bcl-xL, endothelial cells showed a strong angiogenic phenotype in vivo. However, inhibition of Bcl-2 in HDMEC-Bcl-xL with shRNA brought the microvessel density back to levels similar to the ones observed with control HDMEC-LXSN cells. These results suggest that the effect of VEGF secreted in response to Bcl-xL upregulation is potentiated by its ability to induce Bcl-2 expression and subsequent CXCL1 and CXCL8. Otherwise, inhibition of Bcl-2 would not have had such a profound effect on the angiogenic potential of HDMEC-Bcl-xL.
Here, we present data that support a model in which EGF induces expression of Bcl-xL and downstream upregulation of VEGF via activation of MAPK (Figure 8). Upon binding to VEGFR2, VEGF induces upregulation of Bcl-2 expression, which leads to expression and secretion of pro-angiogenic CXC chemokines. These results reveal a novel function for Bcl-xL in a pro-angiogenic signaling pathway. This knowledge suggests a novel therapeutic target for anti-angiogenic cancer therapy that is based on inhibition of Bcl-xL. Since many tumor cells depend on Bcl-xL for survival, such strategy would offer the intriguing possibility of targeting both, the tumor cell and the tumor microvascular network, with the same drug.
Materials and Methods
Plasmids, cells, and ELISA
For transient transfection assays with either pcDNA3 or pcDNA3-Bcl-xL, HDMEC (Cambrex, San Diego, CA, USA) were transfected with 1 μg plasmid for 6 h using Lipofectin (Life Technologies/Invitrogen, Grand Island, NY, USA) according to the manufacturer's instructions. Cells were harvested either immediately after transfection or at 0–24 h post-transfection and supernant was collected for ELISA assays. The generation of endothelial cells stably transduced with Bcl-2, Bcl-xL, Bcl-xL (L130A),16 and Bcl-xL (XB)16 was performed with retroviruses, as described.5, 9 Briefly, the human Bcl-2, Bcl-xL, or mutant Bcl-xL was inserted into the EcoRI cloning site of a retroviral vector (LXSN, gift from D. Miller). The Bcl-2, Bcl-xL, or mutant Bcl-xL construct, or the vector alone, was transfected into PA317 amphotropic packaging cells with Superfect (Qiagen Inc., Valencia, CA). Viral supernatants were collected after 24 h, centrifuged, filtered, and stored at 80°C. HDMEC were transduced with either Bcl-2, Bcl-xL, or LXSN (empty vector) by overnight incubation with one-tenth dilution of the viral supernatant in the presence of 4 mg/ml polybrene (Sigma Chemical Co., St. Louis, MO, USA). Endothelial cell growth medium (EGM2-MV, Cambrex) supplemented with 250 μg/ml G418 (Sigma) was used to select for resistant pools of cells. Bcl-2 and Bcl-xL expressions were confirmed by Western blot analysis. The expression of VEGF, CXCL1, and CXCL8 was evaluated by ELISA (R&D Systems, Minneapolis, MN, USA) and normalized by cell number. For MAPK and NF-κB signaling inhibition assays, chemical inhibitors were used at 0–0.5 μ M I-κB kinase inhibitor peptide (Calbiochem, San Diego, CA, USA) and 0–10 μ M MEK Inhibitor (PD 98059; Calbiochem).
Affymetrix microarrays
About 10 μg of total RNA from HDMEC-Bcl-xL or HDMEC-LXSN were amplified and biotin-labeled according to GeneChip Expression Analysis Technical Manual (Affymetrix, Santa Clara, CA, USA). Fragmented cRNA was hybridized with human gene chip U133A (Affymetrix), chips were washed, and stained with streptavidin R-phycoerythrin (Molecular Probes/Invitrogen, Carlsbad, CA, USA). The chips were scanned and the data were analyzed with Microarray Suite and Data Mining Tool (Affymetrix). The data presented here are representative of microarrays performed with three independent pools of G418-selected cells.
Short hairpin RNA and Western blots
293T cells were transfected with packaging constructs, and either shRNA-Bcl-2, shRNA-Bcl-xL (lentiviral vectors), or control lentiviral vector (shRNA-C; scrambled oligonucleotide sequence), as described.33 Published work was used as a guide to generate short 19-bp hairpins for RNA interference: Bcl-2, nucleotides 500–518 (Genbank M13995)34 and Bcl-xL, nucleotides 714–732 (Genbank NM138578).35 Forty-eight hours post-transfection, virus-enriched supernatant was collected to infect HDMEC cells. Forty-eight hours post-infection of HDMEC with shRNA-containing lentiviruses, supernatant was collected for ELISA and cells were harvested for Western blot analysis. For Western blots, whole cell lysates were resolved by polyacrylamide gel electrophoresis and membranes were probed overnight at 4°C with either a 1 : 10 000 dilution of hamster anti-human Bcl-2 monoclonal antibody (BD Biosciences, San Jose, CA, USA), mouse anti-human Bcl-xL antibody (BD Biosciences), anti-HA (Santa Cruz, Santa Cruz, CA, USA), or anti-flag (Sigma). For MAPK activity assays, cells were starved for 24 h and treated with 0–50 ng/ml EGF, and membranes were probed following the same conditions with either a 1 : 10 000 dilution of rabbit anti-human MAPK-phosphorylated form (Cell Signaling, Danvers, MA, USA), or mouse anti-human MAPK (Cell Signaling). Blots were exposed to appropriate peroxidase-coupled secondary antibodies and washed. Proteins were visualized with enhanced chemiluminescence's detection kit (ECL; Amersham, Piscataway, NJ, USA).
Capillary sprouting assays
About 3 × 104 of endothelial cells were seeded 1.5 ml on type I collagen (Vitrogen 100; Cohesion Technologies, Palo Alto, CA, USA) and cultured in EGM2-MV (Cambrex) supplemented with 50 ng/ml rhVEGF165 for 4 days to induce the capillary sprouting, as described.5 When indicated, cells were exposed to 1 μg/ml monoclonal anti-human CXCR2 antibody (R&D Systems) or to 1 μg/ml mouse anti-IgG2A isotype control (R&D Systems). Alternatively, HDMEC-Bcl-xL, HDMEC-Bcl-xL (L130A), HDMEC-Bcl-xL (XB), HDMEC-Bcl-2, or HDMEC-LXSN were seeded in the collagen and allowed to sprout for 5–7 days. The number of sprouts in six random fields was counted daily in triplicate wells per condition at × 100. At least three independent experiments were performed to confirm reproducibility of data.
SCID mouse model of human angiogenesis
Functional human microvessels were induced in SCID mice, as described.23 Briefly, 1 × 106 endothelial cells were seeded in 6 × 6 × 1 mm poly(L-lactic acid) (PLLA, Medisorb, Cincinnati, OH, USA) biodegradable scaffolds, and two scaffolds were implanted subcutaneously into the dorsum of each SCID mouse (CB.17.SCID, Taconic, Hudson, NY, USA). HDMEC-Bcl-xL cells were infected with lentiviruses expressing shRNA-Bcl-2, and HDMEC were infected with lentiviruses expressing shRNA-Bcl-xL for 48 h before they were harvested and seeded in the scaffolds. Alternatively, endothelial cells infected with control lentiviral vector (shRNA-C), control HDMEC stably transduced with Bcl-2 (HDMEC-Bcl-2), or with empty retroviral vector (HDMEC-LXSN) were seeded in scaffolds and implanted in the mice. All mice were euthanized 14 days after implantation of scaffold-containing cells. The implants were retrieved and fixed in 10% buffered formalin at 4°C overnight, before paraffin embedding.
Immunohistochemistry
Sections were deparaffinized in xylene, rehydrated, and washed in TBS, pH 8.0, then incubated in antigen-retrieval solution (Dakocytomation; Dako, Carpinteria, CA, USA) for 20 min at 95°C. The polyclonal rabbit anti-human factor VIII antibody (Lab Vision Corp., Freemont, CA, USA) was used here to localize the microvascular networks formed by the implanted human endothelial cells, and the immunohistochemistry was performed with Dako EnVision + system kit (AEC, Dakocytomation; Dako), according to the manufacturer's instructions. The number of microvessels in 10 random fields per implant was counted in six scaffolds per condition using light microscope.
Sulforhodamine B assay
About 1 × 103 HDMEC-Bcl-2, HDMEC-Bcl-xL, or HDMEC-LXSN cells were exposed to either 1 μg/ml anti-CXCR2 (R&D Systems) or 1 μg/ml IgG. Alternatively, 4 × 102 HDMEC-Bcl-2, HDMEC-Bcl-xL, or HDMEC-LSXN cells were exposed to lentiviruses encoding shRNA-Bcl-2. After 24–48 h, cells were fixed with 10% trichloroacetic acid, stained with 0.4% SRB (Sigma), and the plates were read in a microplate reader at 565 nm (Tecan, Graz, Austria), as described.8 Triplicate wells per condition were evaluated and the data presented are representative of three independent experiments.
Statistical analysis
Statistical significance was determined at P⩽0.05 by t-tests, or one-way ANOVA followed by the Student–Newman–Keuls post hoc tests using SigmaStat 2.0 software (SPSS, Chicago, IL, USA).
Abbreviations
- HDMEC:
-
human dermal microvascular endothelial cells
- EGF:
-
epithelial growth factor
- VEGF:
-
vascular endothelial growth factor
- CXCR2:
-
CXC receptor-2
- CXCL1:
-
CXC ligand-1
- CXCL8:
-
CXC ligand-8
- NF-κB:
-
nuclear factor-κB
- IκB:
-
inhibitor κB
- MAPK:
-
mitogen-activated protein kinase
References
Reinmuth N, Stoeltzing O, Liu W, Ahmad SA, Jung YD, Fan F et al. Endothelial survival factors as targets for anti-neoplastic therapy. Cancer J 2001; 7: S109–S119.
Nör JE, Polverini PJ . Role of endothelial cell survival and death signals in angiogenesis. Angiogenesis 1999; 3: 101–116.
Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E . Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995; 10: 1024–1028.
Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 1998; 46: 30336–30343.
Nör JE, Christensen J, Mooney DJ, Polverini PJ . Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol 1999; 154: 375–384.
Jain RK, Safabakhsh N, Sckell A, Chen Y, Jiang P, Benjamin L et al. Endothelial cell death, angiogenesis, and microvascular function after castration in an androgen-dependent tumor: role of vascular endothelial growth factor. Proc Natl Acad Sci USA 1998; 18: 10820–10825.
Nör JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ . Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res 2000; 37: 142–150.
Zeitlin BD, Joo E, Dong Z, Warner K, Wang G, Nikolovska-Coleska Z et al. Antiangiogenic effect of TW37, a small-molecule inhibitor of Bcl-2. Cancer Res 2006; 66: 8698–8706.
Nör JE, Peters MC, Christensen JB, Sutorik MM, Linn S, Khan MK et al. Up-regulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res 2001; 61: 2183–2188.
Karl E, Warner K, Zeitlin B, Kaneko T, Wurtzel L, Jin T Chang J et al. Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kappaB and CXC chemokines. Cancer Res 2005; 65: 5063–5069.
Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA et al. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74: 597–608.
Chao DT, Linette GP, Boise LH, White LS, Thompson CB, Korsmeyer SJ . Bcl-XL and Bcl-2 repress a common pathway of cell death. J Exp Med 1995; 3: 821–828.
Chao DT, Korsmeyer SJ . BCL-2 family: regulators of cell death. Annu Rev Immunol 1998; 16: 395–419.
Kedar D, Baker CH, Killion JJ, Dinney CP, Fidler IJ . Blockade of the epidermal growth factor receptor signaling inhibits angiogenesis leading to regression of human renal cell carcinoma growing orthotopically in nude mice. Clin Cancer Res 2002; 8: 3592–3600.
Cui XS, Kim NH . Epidermal growth factor induces Bcl-xL gene expression and reduces apoptosis in porcine parthenotes developing in vitro. Mol Reprod Dev 2003; 66: 273–278.
Minn AJ, Kettlun CS, Liang H, Kelekar A, Vander Heiden MG, Chang BS et al. Bcl-xL regulates apoptosis by heterodimerization-dependent and -independent mechanisms. EMBO J 1999; 18: 632–643.
Baggiolini M, Dewald B, Moser B . Interleukin-8 and related chemotactic cytokines-CXC and CC chemokines. Adv Immunol 1994; 55: 97–179.
Bernardini G, Ribatti D, Spinetti G, Morbidelli L, Ziche M, Santoni A et al. Analysis of the role of chemokines in angiogenesis. J Immunol Methods 2003; 273: 83–101.
Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J et al. PTK787/ZK222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res 2000; 60: 2178–2189.
de Moissac D, Mustapha S, Greenberg AH, Kirshenbaum LA . Bcl-2 activates the transcription factor NFkappaB through the degradation of the cytoplasmic inhibitor IkappaBalpha. J Biol Chem 1998; 273: 23946–23951.
Ricca A, Biroccio A, Del Bufalo D, Mackay AR, Santoni A, Cippitelli M . bcl-2 over-expression enhances NF-kappaB activity and induces mmp-9 transcription in human MCF7 (ADR) breast-cancer cells. Int J Cancer 2000; 86: 188–196.
Distler JH, Hagen C, Hirth A, Muller-Ladner U, Lorenz HM, del Rosso A et al. Bucillamine induces the synthesis of vascular endothelial growth factor dose-dependently in systemic sclerosis fibroblasts via nuclear factor-kappaB and simian virus 40 promoter factor 1 pathways. Mol Pharmacol 2004; 2: 389–399.
Nör JE, Peters MC, Christensen JB, Sutorik MM, Linn S, Khan MK et al. Engineering and characterization of functional human microvessels in immunodeficient mice. Lab Invest 2001; 81: 453–463.
Strieter RM, Polverini PJ, Arenberg DA, Kunkel SL . The role of CXC chemokines as regulators of angiogenesis. Shock 1995; 3: 155–160.
Amin DN, Hida K, Bielenberg DR, Klagsbrun M . Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res 2006; 66: 2173–2180.
Knipling L, Wolff J . Direct interaction of Bcl-2 proteins with tubulin. Biochem Biophys Res Commun 2006; 2: 433–439.
Zachary I, Gliki G . Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res 2001; 3: 568–581.
Shibuya M, Claesson-Welsh L . Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res 2006; 5: 549–560.
Sun C, Hu Y, Liu X, Wu T, Wang Y, He W et al. Resveratrol downregulates the constitutional activation of nuclear factor-kappaB in multiple myeloma cells, leading to suppression of proliferation and invasion, arrest of cell cycle, and induction of apoptosis. Cancer Genet Cytogenet 2006; 1: 9–19.
Jazirehi AR, Vega MI, Chatterjee D, Goodglick L, Bonavida B . Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemosensitization of non-Hodgkin's lymphoma B cells by Rituximab. Cancer Res 2004; 19: 7117–7126.
Coles LS, Lambrusco L, Burrows J, Hunter J, Diamond P, Bert AG et al. Phosphorylation of cold shock domain/Y-box proteins by ERK2 and GSK3beta and repression of the human VEGF promoter. FEBS Lett 2005; 579: 5372–5378.
Kawano Y, Nakamura S, Fukuda J, Sugano T, Takai N, Miyakawa I . The effect of epidermal growth factor on production of vascular endothelial growth factor by amnion-derived (WISH) cells. Growth Factors 2005; 2: 169–175.
Verhaegen M, Bauer JA, Martin de la Vega C, Wang G, Wolter KG, Brenner JC et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res 2006; 66: 11348–11359.
Cioca DP, Aoki Y, Kiyosawa K . RNA interference is a functional pathway with therapeutic potential in human myeloid leukemia cell lines. Cancer Gene Ther 2003; 10: 125–133.
Jiang M, Milner J . Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev 2003; 17: 832–837.
Acknowledgements
We thank Monique Verhaegen for the lentiviral vectors driving shRNAs for Bcl-2 and Bcl-xL, and Craig Thompson for the Bcl-xL mutants L130A and XB. We also thank Chris Yung for his excellent work with the illustration of the model, and Taocong Jin in for his expertise with the Affymetrix microarrays, the Biological Resources Branch (NIH/NCI) for the rhVEGF, and Novartis/Schering for the PTK787/ZK222584 used here. Support for this research was provided by grants R01-DE14601, R01-DE15948, R01-DE16586 from the NIH/NIDCR (JEN); NIH R01 CA107237 (MSS), developmental project grant from the University of Michigan Head & Neck SPORE (JEN); CNPq project 200081–01/2 (EK); and grant PC040286 from the Department of Defense-USA (JEN).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by A Villunger
Rights and permissions
About this article
Cite this article
Karl, E., Zhang, Z., Dong, Z. et al. Unidirectional crosstalk between Bcl-xL and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ 14, 1657–1666 (2007). https://doi.org/10.1038/sj.cdd.4402174
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402174
Keywords
This article is cited by
-
CYD0281, a Bcl-2 BH4 domain antagonist, inhibits tumor angiogenesis and breast cancer tumor growth
BMC Cancer (2023)
-
Tumor microenvironment: an evil nexus promoting aggressive head and neck squamous cell carcinoma and avenue for targeted therapy
Signal Transduction and Targeted Therapy (2021)
-
BCL-XL overexpression promotes tumor progression-associated properties
Cell Death & Disease (2017)
-
Lymphatic endothelial cells support tumor growth in breast cancer
Scientific Reports (2014)
-
BCL2 expression in CD105 positive neoangiogenic cells and tumor progression in angioimmunoblastic T-cell lymphoma
Modern Pathology (2012)
