
Abstract
Rye is a crop with relatively high resistance to biotic and abiotic stresses. However, the resistance to brown rust (Puccinia recondita f. sp. secalis) and pre-harvest sprouting are still not satisfactory. High α-amylase activity is also among the main disadvantages of this species. Therefore, effective tools, e.g. molecular markers, allowing precise and environmentally independent selection of favourable alleles are desirable. In the present study, two kinds of association mapping—genome-wide association mapping (GWAM) based on sequences of DArTSeq markers and candidate gene association mapping (CGAM) based on sequences of ScBx genes—were chosen for development of molecular markers fulfilling these criteria. The analysed population consisted of 149 diverse inbred lines (DILs). Altogether, 67 and 11 single nucleotide polymorphisms (SNPs) identified in, respectively, GWAM and CGAM, were significantly associated with the investigated traits: 2 SNPs with resistance to brown rust, 71 SNPs with resistance to pre-harvest sprouting and 5 SNPs with α-amylase activity in the grain. Fifteen SNPs were stable across all environments. The highest number (13) of environmentally stable SNPs was associated with pre-harvest sprouting resistance. The test employing the Kompetitive Allele Specific PCR method proved the versatility of four markers identified in both GWAM and CGAM.
Similar content being viewed by others

Introduction
Rye (Secale cereale L.) is one of the most important cereals in Eastern, Central and Northern European countries. It was grown on approximately 5.8 million ha worldwide in 2013 [http://faostat.fao.org]. Its importance results both from the unique properties such as low water and soil fertility requirements and good tolerance for biotic and abiotic stresses, good overwintering, high nutritional value connected with the content of dietary fibre and the multipurpose usage—for bread making, animal feed and alcohol production. Recently, it is also gaining attention as a biomass crop (Bushuk 2001). In spite of many advantages, rye exhibits some weaknesses negatively affecting yield quality and quantity, among which the most important are the relatively low resistance to brown rust (Puccinia recondita f. sp. secalis), pre-harvest sprouting (PHS) and high α-amylase activity.
Puccinia recondita f. sp. secalis, an obligate biotrophic basidiomycete fungus, is a commonly occurring and widely distributed major rye pathogen (Roux and Wehling 2010). A rust epidemic results in premature leaf senescence and in consequence a reduced number of florets in the spike. The disease intensity is correlated with reduction of kernel number, yield quality and quantity. Available estimates indicated 11 to 27% thousand kernel weight reduction (Miedaner and Sperling 1995) and yield decrease of up to 80% (Solodukhina 1997 2002) depending on genotype. Modern rye varieties do not always exhibit a satisfactory level of resistance to brown rust (Solodukhina 1997; Miedaner et al. 2002; Roux et al. 2004). This problem, in particular, concerns the population varieties which dominate in Poland. Wehling et al. (2003) identified in rye two dominant genes, Pr1 and Pr2, both conferring resistance through hypersensitivity, which were recognized as active against a local population of P. recondita as well as isolates of the fungus obtained from single pustules and representing various virulence. Three additional genes, Pr3, Pr4, Pr5, associated with race-specific resistance to brown rust, were identified by Roux et al. (2004). Specific resistance is an effective strategy, often used in breeding, against P. recondita as well as other biotrophic pathogens, but unfortunately its durability is not satisfactory (Miedaner et al. 2012). An alternate approach focuses on nonspecific resistance. It is also known as non-isolate-specific or partial resistance as the disease development is reduced but not completely stopped.
So far, nonspecific resistance genes coding for ABC transporter and proline-containing protein have been recognized in wheat (Krattinger et al. 2009) and in rice (Fukuoka et al. 2009). The concept of nonspecific resistance against brown rust of rye has not been studied in depth.
PHS is the phenomenon occurring during the later stages of cereal grain development when prolonged rain induces premature germination whilst the grain is still in the ear. Water absorbed by the rye kernel activates enzymes, especially α-amylase, which causes degradation of the endosperm starch and a significant reduction of grain quality. Germinated seeds represent material of low sowing and technological value. PHS has been considered to be one of the main factors negatively affecting the yield and quality of crops grown in wet or humid weather conditions. PHS resistance is a complex quantitative trait, controlled by a number of quantitative trait loci (QTL) mapped on each of the seven rye chromosomes (Masojć and Milczarski 2009; Masojć et al. 2009; Myśków et al. 2012). Studies of QTL controlling pre-harvest sprouting and α-amylase activity in rye demonstrated that these two complex genetic systems coincide to a great extent (Masojć and Milczarski 2009; Myśków et al. 2012).
Increasing resistance to rust, PHS and lowering of α-amylase activity are still current objectives in breeding of rye varieties. Finding molecular markers for these agronomic traits is therefore essential, since application of novel molecular tools in breeding will speed up selection of valuable genotypes.
Association mapping, also called linkage disequilibrium mapping, involves searching for genotype (usually individual single nucleotide polymorphisms (SNPs) or SNP haplotypes)—phenotype correlations in unrelated individuals using dedicated statistical methods (Abdurakhmonov and Abdukarimov 2008; Zhu et al. 2008; Rafalski 2010). The association mapping approach creates possibilities to generate good quality markers for marker-assisted selection (MAS). The most effective are functional markers reflecting gene polymorphism tightly linked with the trait which is a direct cause of phenotypic variation. Association mapping provides opportunities to find such markers in a broad spectrum of genetic resources. Its potential results from the likelihood of higher mapping resolution because of the utilization of more recombination events in the germplasm’s developmental history (Abdurakhmonov and Abdukarimov 2008; Rafalski 2010). Thus, association mapping has become a promising approach compared to traditional linkage mapping. There are two main types of association mapping: genome-wide association mapping (GWAM) and candidate gene association mapping (CGAM). The GWAM approach surveys genetic variation in the whole genome to find signals of association for various complex traits, whereas CGAM correlates DNA polymorphisms in selected candidate genes and the trait of interest (Zhu et al. 2008; Rafalski 2010).
In our research, DArTSeq markers were used for GWAM and ScBx genes for CGAM. DArTseq technology utilizes the DArT marker platform in combination with next-generation sequencing platforms and represents a new implementation of sequencing of complexity-reduced representations (Sansaloni et al. 2011; Courtois et al. 2013). ScBx1–5 genes, recently sequenced and characterized by Bakera et al. (2015), control the five steps of benzoxazinoid (BX) biosynthesis (starting with the conversion of indole-3-glycerol phosphate to indole followed by four monooxidations with the final product 2,4-dihydroxy-1,4-benzoxazin-3-one). BXs are protective and allelopathic secondary metabolites are found in a large number of species belonging to the Poaceae family, including the major agricultural cereals maize, wheat and rye (Frey et al. 2009; Niemeyer 2009). Several studies have shown a relationship between BX and disease resistance (Niemeyer 2009). BXs have also been shown to play a role in the inhibition of gibberellin-induced α-amylase activity in barley seeds (Kato-Noguchi 2008). Thus, genes encoding enzymes participating in BX biosynthesis related enzymes seem to be good candidates for association mapping.
In cereals, mainly in wheat, there are many examples of successful application of association analysis for mapping QTL controlling PHS resistance (Lohwasser et al. 2013; Albrecht et al. 2015), rust resistance (Philomin et al. 2015; Kertho et al. 2015), α-amylase activity (Emebiri et al. 2010) or frost tolerance (Li et al. 2011).
So far, a relatively large number of molecular markers have been developed for rye: RAPD (Masojć et al. 2001; Bednarek et al. 2003), RFLP (Korzun et al. 2001; Masojć et al. 2001), AFLP (Bednarek et al. 2003), SSR (Saal and Wricke 1999; Khlestkina et al. 2004), DarT (Bolibok-Brągoszewska et al. 2009; Milczarski et al. 2011), GBS (Milczarski et al. 2016), and there are many examples of their successful use for tagging certain traits, e.g. PHS (Masojć and Milczarski 2009; Masojć et al. 2009; Myśków et al. 2012), aluminium tolerance (Gallego et al. 1998) or frost tolerance (Li et al. 2011). Nevertheless, no attempts have been made to perform association analysis of PHS, rust resistance and α-amylase activity nor have DArTSeq markers been used for such studies in rye yet.
The aims of the present study were the following: (1) to characterize a broad collection of rye diverse inbred lines representing the variability among Polish breeding materials with respect to brown rust resistance, pre-harvest sprouting and α-amylase activity, (2) to identify SNPs in ScBx genes and DArTSeq markers associated with these traits.
Materials and Methods
Plant Material
The plant material used in the experiments consisted of 153 diverse inbred lines (DILs) bred in two Polish breeding companies—Danko Plant Breeders Ltd. (40 DILs, designated as D1–D40) and Poznań Plant Breeders Ltd. (30 lines, designated as P1–P30); Botanical Garden of Polish Academy of Science (8 lines; OG1–OG8); Warsaw University of Life Sciences, Department of Plant Genetics, Breeding and Biotechnology (5 lines designated as W1–W5); West Pomeranian University of Technology, Department of Plant Genetics, Breeding and Biotechnology (20 lines designated as S1–S20); and Wrocław University of Environmental and Life Sciences, Department of Genetics, Plant Breeding and Seed Production (50 lines designated as WR1–WR50).
In 2013, 103 lines were tested, and the set was increased to 153 lines in 2014.
The name W1 is a synonym of L318. Additional plant material, not related (but belonging to the same Petkus germplasm pool) to DILs used in the association analysis, consisting of 60 recombinant inbred lines (RIL) from the mapping population 541 × Ot1-3 (Milczarski et al. 2007) and 20 inbred lines from Danko Plant Breeders Ltd. was used in the Kompetitive allele-specific PCR (KASP) analysis. The inbreeding degree of DILs was at least S5 (usually higher), except for WR lines, for which it was S4.
Methods
Phenotyping
Each line was represented by 33 plants per location and year, grown in a randomized block design (11 plants per replicate). The experiments were performed in two seasons, 2013 and 2014, in two locations: an experimental field of the West Pomeranian University of Technology in Szczecin (N 53° 26′ 51.291″, E 14° 31′ 42.1278″) and Danko Plant Breeders Ltd. in Choryń (N 52° 02′ 19.7″, E 16° 46′ 09.0″). Together, observations from four experiments designated as WPUT 2013, WPUT 2014, DANKO 2013 and DANKO 2014 were scored.
The following traits of DILs were phenotyped: brown rust resistance (R-R), pre-harvest sprouting resistance (PHS-R) and α-amylase activity in grain (AMY).
R-R
The reaction of DILs to P. recondita was examined in field conditions where the source of inoculum was uredospores of a local population of the fungus, naturally occurring in the environment and spontaneously infecting the plants. No special treatments were applied for enhancing/increasing the infection potential of the fungus. For comparison purposes, the set of tested lines was supplemented with two rye cultivars, Dańkowskie Złote as a susceptibility reference and Bosmo as a tolerance reference.
Rating was performed at maximum brown rust epidemic intensity and based on average evaluation of all plants in the plot.
Plant infection level was evaluated according to a six-degree scale:
-
0—resistant, no symptoms on plants
-
1—resistant, chlorosis/necrosis and/or single uredinia sporadically visible on leaves
-
2—moderate resistant; single small uredinia regularly present on majority of plants
-
3—moderately susceptible; up to 40% of leaf surface covered with uredinia
-
4—susceptible; up to 70% of leaf surface covered with uredinia
-
5—very susceptible; over 70% of leaf surface covered with uredinia.
PHS-R
Three to six spikes from each DIL and each replicate were harvested at full ripeness and kept at room temperature for 3 days. Then, the spikes were sprayed with water for 5 min each day and kept in a moisture chamber at 20 °C for a period of 7 days. Resistance to PHS was determined as a percentage of germinated kernels (the lower the percentage, the higher the resistance).
AMY
1.5 g of kernels with no visible signs of sprouting from each DIL and each replicate was milled into flour. Grain used for the α-amylase assay was stored at room temperature for the first 2 weeks after harvest and then kept at 4 °C for the few months before analysis. For α-amylase extraction, 1 g of flour was mixed with 4 ml of distilled water, equilibrated for 10 min at room temperature and finally centrifuged at 8000 rpm at 4 °C for 20 min. Alpha-amylase activity in mature grain was detected using a simple gel diffusion method (Masojć and Larsson-Raźnikiewicz 1991) where the diameter of the diffusion circle shows a linear relationship with the logarithm of the enzyme activity given in units per milliliter. A calibration curve was obtained by a series of dilutions of a commercial alpha-amylase from barley malt (Sigma).
The number of lines phenotyped in terms of PHS-R and AMY was slightly lower than the number of lines evaluated in respect of R-R in some experiments (Table 1) because of plant losses before the spike harvest began. In two trials (DANKO 2013 and WPUT 2014), one and three RILs, correspondingly, had delayed vegetation, and because of this, they were not evaluated in terms of R-R.
Genotyping and SNP Data Processing
Genotypic data for association mapping came from polymorphisms identified in DArT and candidate gene sequences.
DArT Sequences
One hundred forty-eight DILs were genotyped. The preparation for DArTSeq genotyping was performed as described by Bolibok-Brągoszewska et al. (2009). Briefly, DNA was isolated from bulked (eight plants per each DIL), lyophilised two-week-old leaves using the CTAB method (Murray and Thompson 1980). DNA purity and concentration were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, USA). DArTseq genotyping was done in the Diversity Arrays Technology Pty. Ltd., Australia, using the platform developed by Cruz et al. (2013). SNPs were extracted with the accompanying metadata (including average marker reproducibility calculated from technical replications). For the association analysis, only DArT sequences meeting the following criteria were selected: one SNP within a given sequence (69 nt), minor allele frequency (MAF) > 0.25 and the missing observation fractions <10%. SNP sequences were mapped in Sce assembly 02 (Haseneyer et al. 2011) using the Blast service at https://www.gabipd.org/ with default parameters.
ScBx Genes
ScBx1-5 genes (comprising exons, introns, 3′UTRs and promoters, except for ScBx1 in which no 3′UTRs were assayed; Table SI, Electronic Supplementary Material, ESM), previously sequenced and characterized in rye inbred line L318 (Bakera et al. 2015), were re-sequenced in a set of 143 DILs using the Roche 454 sequencing method (Genomed S.A., Warsaw). The smaller number of DILs included in the analysis than the total number of phenotyped lines resulted from the inability to obtain the common fragment of the 3′UTR of the ScBx5 gene, in five lines. The Roche 454 paired end reads (median read length = 190 nt, median number of reads per DIL = 320,587, min = 900, max = 7,211,998) were mapped to the reference sequence using Bowtie 2 (Langmead et al. 2009), allowing for a maximum of two mismatches. The mapping results were processed by the SNP calling pipeline based on Samtools (mpileup) and Bcftools (index, call) software (Li et al. 2009; Li 2011, software online documentation).
The alleles the same as present in the reference line L318 were marked as REF, and the alleles resulting from a given SNP were marked as ALT.
Statistical Analysis and Association Mapping
Data were analysed by ANOVA in a model with fixed effects of years (Y), locations (L), and of Y × L interaction, and random effects of DILs and DIL × (Y, L) interactions. Association mapping, based on SNP data and on mean values of traits obtained in individual experiments, was done using the method allowing for interaction of genetic effects with the environment developed by van Eeuwijk et al. (2010) and Malosetti et al. (2013), based on the mixed linear model with the population structure estimated by eigenanalysis (principal component analysis applied to all markers). The compound symmetry variance-covariance model was used for environmental variation. All analyses and visualizations of results were done in GenStat 17 (EVSN Int. 2013). Significance of associations between traits and DArTseq markers was assessed on the basis of P values corrected for multiple testing by the Benjamini-Hochberg method as implemented in R function p.adjust.
Mapping DArTSeq Markers
The chromosome position of DArTSeq markers significantly associated with investigated traits at P < 0.001 was analysed based on a genetic map of population S (Milczarski et al. 2011) which was extended with DArTseq markers (Milczarski et al. 2016). This S map was a reference. For the SNPs, which could not be localized on the S map, other genetic maps were used: a genetic map based on population H (Milczarski et al. 2011) extended with DArTseq markers (Bolibok-Brągoszewska, personal communication) and a de novo constructed map based on population 541 × 723 comprising DArTseq markers (Milczarski et al. personal communication). The positions of SNPs (verified through ID number and sequence) on these maps were recalculated in relation to the reference map. For the estimation of position, SNP shared markers from compared maps were used.
For the construction of genetic maps, the program JoinMap 4.0 with the ML (maximum likelihood) mapping algorithm was applied.
Evaluation of Associations
Selected SNPs found to be significantly and stably associated with R-R and PHS-R were evaluated using the KASP assays (LGC Genomics, Herts, England). An additional selection criterion was the ability to design good quality primers for the analysed sequence. The 50 bp sequences, each upstream and downstream around the SNP, were the templates for design of primer pairs. KASP reactions were run in 10 μl reactions including 5 μl of KASP master mix, 0.14 μl of primer mix (LGC Genomics, Herts, England), and 5 μl of 17–20 ng/μl genomic DNA. PCR and fluorescent endpoint readings were carried out using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Altogether, 120 lines were genotyped: 40 DILs included in the association analysis (experimental set, EXS)—20 DILs in the experiment on SNPs associated with PHS-R and 20 DILs in the experiment on SNPs associated with R-R—and 80 lines not included in the association analysis (evaluation set—EVS), comprising 60 recombinant inbred lines (40 taken for the experiment on SNPs associated with PHS-R and 20 for those associated with R-R) and 20 inbred lines (taken for the experiment on SNPs associated with R-R) phenotyped previously by Masojć et al. (2009) and by breeders from the Danko Plant Breeders Ltd., respectively. The lines for KASP test were chosen randomly from extreme groups of phenotypes. Each subset was composed in half of resistant and in half of susceptible lines, but the phenotypes were not revealed until completing KASP analysis.
Results
Phenotyping
The comparison of R-R, PHS-R and AMY levels in all experiments is shown in Table 1. The highest resistance to brown rust was observed in WPUT 2014, whereas the lowest R-R value was observed in DANKO 2014. Oppositely, for PHS, the highest resistance was observed in DANKO 2014 and the lowest one in WPUT 2014. The lowest activity of AMY was also noted in the DANKO 2014 trial whilst the highest AMY enzymatic activity was noted in the same location but in 2013. Results of ANOVA (Table SII, ESM) demonstrated that for all traits, the differences between years, locations and the year × location interaction were significant. Comparison of variance components among themselves and with their standard errors showed that the component for DIL genotypes dominates the interaction component for R-R, whereas for PHS-R, the component for interaction of DIL genotypes with locations was the largest.
Correlations of observed trait levels were not significant (Table 2) except for AMY and R-R, which were correlated in one experiment—WPUT 2013. The interactions of lines with years were larger—in relation to their standard errors—than the interactions with locations. The relationships between variance components are reflected in the estimates of broad-sense heritability, with the largest value for R-R (Table SII, ESM). Phenotypic and genetic correlations between environments (Table 3) confirm a stronger relationship between locations than between years for R-R and PHS-R, and an outlying behaviour of observations in WPUT 2013 for AMY.
The groups of lines provided by different breeders differed with respect to mean R-R and AMY, whereas for PHS-R, the differences were not significant (Table 4). Lines D had the significantly lowest mean level of R-R, and lines S had the significantly highest level of AMY.
Genotyping Data and Population Structure
DArTSeq
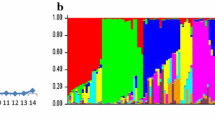
Out of 36,894 polymorphisms obtained, 2330 SNPs were selected for GWAM using the criteria specified above. Population structure estimated by eigenanalysis (Fig. 1a) shows large subsets of D and P lines (provided by breeding companies) differentiated from the others. The rest of DILs from these two groups were similar to OG, S and W lines, and form the central cluster in the graph. The WR lines were also separated from the others; their homozygosity (median 0.75; from 0.51 to 0.90) was lower than that of the rest of the lines (median 0.98; from 0.62 to 0.99). By performing the χ (Albrecht et al. 2015) independence test for origin of lines and genotypic classes for SNPs (details not shown), SNP 3598261 was identified as explaining well the dispersion of lines D (mostly G/G) and P (mostly A/A) (Fig. 1b). The fraction of homozygous observations for individual SNPs was from 0.53 to 0.97.

Population structure estimated by eigenanalysis
ScBx Genes
In the sequences comprising full ScBx genes and their promoters, 70 SNPs were identified in a set of 143 DILs—26, 22, 1, 7 and 14 in ScBx1, ScBx2, ScBx3, ScBx4 and ScBx5 genes, respectively. All 70 polymorphisms were included in CGAM. Homozygosity of DIL with respect to these SNPs ranged from 0.28 to 1.00. There were six SNPs with the fraction of homozygous observations less than 0.65 (Fig. 2). In general, the SNPs located in ScBx5 were mostly heterozygous. The population structure estimated by eigenanalysis (Fig. 1c) shows three separate groups of lines; however, the dispersion of lines does not exhibit any relationship to breeding groups. The population structure was determined mostly by the genotypes observed in genes ScBx1 and ScBx2, for example, by a combined genotype at SNPs ScBx1_4491 and ScBx2_212 (Fig. 1d).

Homozygosity for SNP
Association Mapping: CGAM, GWAM
GWAM
The Q-Q plots of estimated allelic effects for all SNPs (Fig. S1, ESM) show their approximate normality and relatively large size of the extreme effects, especially for PHS-R. Based on the mixed linear model analysis, the number of SNPs with associations with investigated traits significant at FDR < 0.05 in GWAM was 67 (Table SIII, ESM). For R-R, one significant association was found (marker 3363612) with a negative (favourable) effect of the alternative allele. For PHS-R, 61 associations were detected (7 stable across all environments). Four associations that were stable over environments had a negative (favourable) effect of the alternative allele. For AMY, there were five significant associations, one with positive effects stable over environments. For PHS-R, for the associations with an interaction, the smallest allelic substitution effects were observed in almost all cases in WPUT 2013, and the largest in DANKO 2013; the mean allelic effect over environments was negative (favourable) in 35 cases and positive (unfavourable) in 23. All SNPs in DArT sequences significantly and stably associated with the traits of interest are listed in Table 5.
Out of 67 SNPs significantly associated with the investigated traits, 35 were mapped on the reference map. Additionally, five DArTSeq markers were mapped to Sce assembly 02 and one marker (3602388) to 2R, 110.7 cM in the map of Martis et al. (2013) (Table 5; Table SIII, Fig. S2, ESM). Among eight markers which had effects stable across environments, only two DArTSeq markers—5200240 and 7104304—remained unmapped.
CGAM
The number of SNPs associated with investigated traits significant at P < 0.01 was 11 (Table SIV, Fig. S1, ESM), with 1 and 10 for R-R and PHS-R, respectively. Six of these associations were identified in the ScBx1 gene. The polymorphism associated with R-R was found in the gene ScBx4 (in the first intron). Polymorphisms associated with PHS-R were detected in ScBx1 (six SNPs: two in the promoter sequence, two in the seventh exon and two in the 3′UTR region), ScBx2 (three SNPs: two in the first exon and one in the second exon) and ScBx4 (one SNP in the first intron).
The SNP associated with R-R had an effect stable over all experiments. For PHS-R, 6 out of 10 of the associations were stable across all environments. All associations exhibited a positive effect of the ALT allele (resulting from a given SNP) in comparison to the REF allele (the same as present in line L318). No ScBx polymorphism was found to be associated with AMY activity.
SNPs significantly and stably associated with the traits of interest (Table 5) were located in the promoter and 3′UTR regulatory sequences of the ScBx1 gene, predominantly. No SNP associated with the analysed traits was detected in the ScBx3 gene.
Evaluation of Associations
To verify whether the SNPs associated with the investigated traits of DILs are also associated with a given phenotype in the unrelated rye inbreds, KASP analysis was performed. Out of 16 SNPs significantly and stably associated with the investigated traits, four SNP markers were chosen for KASP analysis: ScBx5_1593 associated with R-R, 3602388, ScBx1_1367 and ScBx1_4515 associated with PHS-R. The marker ScBx5_1593 was associated with R-R only at P < 0.05, but it was included in the analysis as stable, with a large favourable effect.
All markers, both those associated with R-R and those related to PHS-R, identified the phenotype with correctness higher than 86%. Usually, a slightly higher percentage of mismatches (i.e. other phenotype than expected taking into account the results of association analysis) was noted in the case of the experimental set (Table 6). Only moderate resistance to brown rust (evaluation score under 2.5) associated with ALT GG homozygosity was identified better in the experimental set than in the evaluation set.
Discussion
In this study, 78 SNPs associated with three rye traits—resistance to brown rust and pre-harvest sprouting, and α-amylase activity—were identified; nearly 20% of associations were stable over environments.
Correlation Between Studied Traits
Out of the three investigated traits, two—R-R and AMY—were found to be weakly correlated in one of the experiments. Although the common mechanisms determining these two traits are unknown, there is published evidence that genes/QTL controlling them are linked. Emebiri et al. (2010) showed that in hexaploid wheat, the gene cluster for rust resistance (Lr37/Sr38/Y17) on chromosome 2A is significantly associated with late maturity α-amylase (LMA). It can be assumed that the gibberellins (GA), phytohormones that regulate many common processes throughout the plant life cycle, are the common factor linking these two traits. De Vleesschauwer et al. (2012) reported that treatment of wild-type Nipponbare rice with increasing concentrations of GA3 enhanced resistance to P. graminicola in a concentration-dependent manner. Conversely, a decrease of endogenous GA levels using the GA biosynthesis inhibitor uniconazole (Izumi et al. 1984) promoted disease susceptibility. The relationship between gibberellin and α-amylase biosynthesis in rice has been reported by Kaneko et al. (2002), who showed that 3-beta-hydroxylase produced by the OsGA3ox2 gene is important for induction of expression of the RAmy1A gene encoding α-amylase. They concluded that the synthesis of active GA in the epithelium is important for α-amylase expression in the endosperm.
Our results show that in the case of another pair of traits—PHS-R and AMY—there is no correlation. A similar conclusion was drawn following studies on biparental rye populations (Twardowska et al. 2005). Although, as shown earlier, α-amylase activity in rye grain and predisposition to pre-harvest sprouting have many, partially overlapping, QTL, their expression may be regulated by different mechanisms (Masojć and Milczarski 2009; Masojć et al. 2011). The independent genetic control of these traits was also reported for wheat (Mares and Mrva 2014). This information is very important for rye breeders, since in view of our results, AMY and PHS resistance should be considered as independent traits in selection.
Association Analysis
To identify SNPs associated with three traits of rye—resistance to brown rust and pre-harvest sprouting, and activity of α-amylase—two approaches were selected: GWAM based on sequences of DArTSeq markers and CGAM based on polymorphism in ScBx genes. We assumed that this is the most effective tool to generate effective molecular markers for these traits. Its potential results from the likelihood of higher mapping resolution because of the utilization of more recombination events in the germplasm’s developmental history (Abdurakhmonov and Abdukarimov 2008). The success of association mapping depends on many factors, among which, the flowering biology is one of the most important (Abdurakhmonov and Abdukarimov 2008). Rye is an open pollinated species with a majority of heterozygous loci. It may decrease LD and result in low precision of association mapping. To overcome this drawback, it is suggested to use a synthetic population like nested association mapping (NAM) in maize (Yu et al. 2008). The plant materials used in our study consisted of a set of diverse inbred lines, mostly highly homozygous. The choice of such a population is, however, a compromise as, on the one hand, it increased mapping precision, and on the other, inbred lines of rye always suffer from heterozygosity. The association analysis method that was used allows for heterozygosity of genotypes, as it relies on the regression of phenotypic values on genetic predictors computed from the numbers of given alleles for SNPs that can take values 0, 1 or 2 (van Eeuwijk et al. 2010).
The population structure, as described by means of eigenanalysis of marker data, was related to the origin of lines in the case of one of the DArTSeq markers; it was shown to differentiate the groups provided by two breeding companies, but it was not unique, and a number of SNPs provided a similar explanation. The structure resulting from polymorphisms in ScBx genes had no obvious relation to breeding groups. The sequences of Bx genes were never a target of selection in rye breeding programmes.
Genome-Wide Association Mapping
For genome-wide association mapping, we decided to use one of the genotyping by sequencing methods, DArTSeq technology, because of its unquestionable competitiveness and efficiency. DArTSeq technology enables selection of the genome fraction corresponding predominantly to gene sequences, which is based on the use of a combination of restriction enzymes PstI/TaqI. These enzymes separate low copy sequences (most informative for marker discovery and genotyping) from the repetitive fraction of the genome and use the next-generation sequencing platforms (Sansaloni et al. 2011; Courtois et al. 2013). This technology is therefore positioned in the area of high resolution mapping and detailed genetic dissection of traits. Genotyping by sequencing in plants has been recently effectively employed in marker-trait association analysis for e.g. root traits in rice (Courtois et al. 2013), resistance to Puccinia hordei in barley (Dracatos et al. 2014) and complex disease resistance traits in bread wheat (Li et al. 2015). To find significant SNP-trait associations in GWAM, we used a statistical method based on a mixed linear model, in which the SNP effect is modelled by regression of the trait on the number of one of the alleles in the genotype. The analysis is based on data from all environments and takes into account the possibility that the regression coefficient is different in different environments, which is taken as evidence of the “allelic effect × environment interaction”. In case of no interaction, the common allelic effect is estimated. The advantage of the method used is that it allows for formal statistical confirmation of significant interactions, unlike the approaches based on separate data analysis in consecutive environments.
In our study, we identified 67 SNPs in DArT sequences that were linked to observed traits, predominantly associated with PHS-R. The majority of identified trait-SNP relationships were affected by the environment. It is worth emphasizing that the climate conditions in June and July, in two consecutive seasons (2013 and 2014), were notably different. For example, in June 2013, the temperatures were extremely high and relatively stable, whereas in June 2014, significant temperature fluctuations were observed. In Szczecin, in July 2014, there was drought and in Choryń, the rainfall in this month was much higher than average. June and July are critical months for the expression of the analysed traits. Nevertheless, the trait relationship with eight SNPs (12%)—seven for PHS-R and one for AMY—turned out to be stable across all environmental conditions represented by two locations and two consecutive seasons.
More than 50% of SNPs in DArT sequences associated with the investigated traits have been mapped on maps constructed previously (Milczarski et al. 2011, 2016), the majority of them on chromosomes 6R and 7R. Map positions of numerous SNPs related to PHS-R and AMY coincide with QTL detected for these traits by Masojć and Milczarski (2009) and by Masojć et al. (2009) using biparental populations. SNPs associated with R-R have been mapped on chromosome 1R to the same region as genes Pr3, Pr4 and Pr5 (Roux et al. 2004).
Candidate Gene Association Mapping
For CGAM, ScBx genes, isolated and sequenced recently in our group (Bakera et al. 2015), which control the biosynthesis of BXs, seemed to be perfect candidates. As it was underlined by Zhu et al. (2008), CGAM could be an attractive, rapid and inexpensive approach for identifying markers associated with genes/quantitative trait loci (QTL) for various quantitative traits, provided that the function and sequence of candidate genes are known. BXs have been shown to have various unique properties—from allelopathic to human health-beneficial ones (Makowska et al. 2015). The resistance to fungal pathogens and the activity of α-amylase are listed among the features dependent on BX (for review see Niemeyer 2009). In the present study, 11 environmentally stable SNPs found in ScBx genes were associated with either R-R (1 SNP) or PHS-R (10 SNPs). Three SNPs—ScBx1_4515, ScBx4_1583 and ScBx4_1627—were previously shown to be associated with BX content in the above ground parts of plants (the first of the listed markers) and roots (the remaining two markers) (Rakoczy-Trojanowska et al. 2016). Most of the identified SNPs were positioned in the promoters, which could be connected with gene expression. According to Zhu et al. (2008), promoter, intron, exon and 5′/3′-untranslated regions are all reasonable targets for identifying candidate gene SNPs, with non-coding regions expected to have higher levels of nucleotide diversity than coding regions. For example, Liu et al. (2013) showed that the polymorphisms identified in the promoter of the ZmDREB2.7 gene were associated with drought tolerance among maize varieties. The authors suggested that these polymorphisms may influence gene expression in response to drought stress.
Nearly 80% of SNPs were located in the ScBx1 and ScBx2 genes and more than half of them in the promoters. It can be assumed that the expression difference of ScBx1 and ScBx2 genes causes differences in levels of BX content and, consequently, the levels of traits potentially dependent on HAs. The ScBx1 and ScBx2 genes are thought to be the most crucial genes for the whole BX biosynthesis pathway, as ScBx1 controls the first step, a branch point in BX biosynthesis (the conversion of indole-3-glycerol phosphate to indole, which is then delivered to the monooxygenation reaction), and ScBx2 is responsible for the synthesis of the first hydroxamic acid: indolin-2-on (Makowska et al. 2015). A significant positive correlation between the expression level of the ScBx1 gene and the content of BXs in young rye plants has been shown by Groszyk et al. (2013, 2015). La Hovary (2012) found that BX1 and BX2 transcript levels paralleled the concentrations of BXs.
SNPs stably associated with the traits of interest usually were present in non-coding gene regions (introns of ScB4 and ScBx5, 3′UTRs of ScBx1) and 3′ regulatory sequences (promoters of ScBx1). These polymorphisms can modify protein properties, at least, or impair enzyme synthesis. Bakera et al. (2015) showed that the changes in BX2 and BX4 proteins (the presence of unique α-helices) resulted from indels present in ScBx2 and ScBx4 genes, respectively.
Evaluation of Identified SNPs
In contrast to the results of GWAM, in the case of CGAM, most (64%) associations were not affected by the environment. Altogether, we identified 15 environmentally stable associations with SNPs (8 in DArT sequences and 7 in ScBx genes). All these polymorphisms may probably serve as a template for developing effective molecular markers for MAS. As stated above, the weather in summer, in two consecutive seasons, was considerably different. These circumstances show the potential of the detected SNPs. To test the reliability, we performed a validation experiment by applying the SNPs in an independent set of accessions not included in the association analysis (designated as the evaluation set) using the KASP method. This analytical approach is agreed to be a cost-effective and accurate marker assay, especially for genotyping large segregating populations and in the case of numerous samples with a few SNPs (Saxena et al. 2012; Semagn et al. 2014). There are many publications relating either to the technology itself or to its application in crop improvement programmes, e.g. in maize (Ertiro et al. 2015), wheat (Bernardo et al. 2015) or rice (Liu et al. 2016). However, none of them applies rye.
As expected, KASP turned out to be sufficiently sensitive to differentiate between both types of homozygotes and heterozygotes. The precision as well as relative simplicity, cost-effectiveness and quick analysis make KASP worth recommending as a technology in pre-breeding works. Generally, in the evaluation set, a given phenotype correlated with a given allele. Nevertheless, minor mismatches (at most 13.33% mistakenly identified phenotypes) were observed. They might be caused by insufficiently accurate phenotyping in single cases rather than by the genetic determinants. Nevertheless, the accuracy of genotype-based selection at a level higher than 86% may be of interest to breeders, especially as the evaluation set consisted of lines which were not included in the association mapping and were not related by pedigree with the experimental set of DILs, even when they were bred in the same institution or company. Of course, lines in EVS were not entirely unrelated to DILs, as all plant material used in our study belongs to the same Petkus germplasm pool.
Moreover, the obtained results, especially KASP-evaluated markers for dominantly inherited R-R, can be almost directly implemented in practice, e.g. in marker-assisted selection, because of the structure of the analysed population consisting of inbred lines. Nearly 50% of DILs come from the breeding programmes of two Polish breeding companies. As pointed out by Begum et al. (2015), performing association mapping on a panel of adapted breeding lines rather than on a diversity panel provides an opportunity to apply the results directly to breeding programmes.
References
Abdurakhmonov I, Abdukarimov A (2008) Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int J Plant Genomics 2008:574927
Albrecht T, Oberforster M, Kempf H et al (2015) Genome-wide association mapping of preharvest sprouting resistance in a diversity panel of European winter wheats. J Appl Genet 56(3):277–285
Bakera B, Makowska B, Groszyk J et al (2015) Structural characteristics of ScBx genes controlling the biosynthesis of hydroxamic acids in rye (Secale cereale L.) J Appl Genet 56(3):287–298
Bednarek PT, Masojć P, Lewandowska R, Myśków B (2003) Saturating rye genomic map with amplified fragment length polymorphism (AFLP) and random amplified polymorphic DNA (RAPD) markers. J Appl Genet 44:21–33
Begum H, Spindel JE, Lalusin A et al (2015) Genome-wide association mapping for yield and other agronomic traits in an elite breeding population of tropical rice (Oryza sativa). PLoS One 10(3):e0119873
Bernardo A, Wang S, St Amand P, Bai G (2015) Using next generation sequencing for multiplexed trait-linked markers in wheat. PLoS One 10(12):e0143890
Bolibok-Brągoszewska H, Heller-Uszyńska K, Wenzl P et al (2009) DArT markers for the rye genome—genetic diversity and mapping. BMC Genomics 10:578
Bushuk W (2001) Rye production and uses worldwide. Adapted from a chapter by the author in rye: pro-duction, chemistry, and technology, 2nd ed., published by AACC, St. Paul, MN, 2001 46(2):71–73
Courtois B, Audebert A, Dardou A et al (2013) Genome-wide association mapping of root traits in a japonica rice panel. PLoS One 8:e78037
Cruz VMV, Kilian A, Dierig DA (2013) Development of DArT marker platforms and genetic diversity assessment of the US collection of the new oilseed crop lesquerella and related species. PLoS One 8:e64062
De Vleesschauwer D, Van Buyten E, Satoh K et al (2012) Brassinosteroids antagonize gibberellin- and salicylate-mediated root immunity in rice. Plant Physiol 158(4):1833–1846
Dracatos PM, Khatkar MS, Singh D, Park RF (2014) Genetic mapping of a new race specific resistance allele effective to Puccinia hordei at the Rph9/Rph12 locus on chromosome 5HL in barley. BMC Plant Biol 14:1598
van Eeuwijk FA, Bink MCAM, Chenu K, Chapman SC (2010) Detection and use of QTL for complex traits in multiple environments. Curr Opin Plant Biol 13:193–205
Emebiri LC, Oliver JR, Mrva K, Mares D (2010) Association mapping of late maturity α-amylase (LMA) activity in a collection of synthetic hexaploid wheat. Mol Breeding 26:39–49
Ertiro BT, Ogugo V, Worku M, Das B, Olsen M, Labuschagne M, Semagn K (2015) Comparison of Kompetitive allele specific PCR (KASP) and genotyping by sequencing (GBS) for quality control analysis in maize. BMC Genomics 16:908
EVSN International (2013) GenStat for Windows 16th Edition EVSN International, Hemel Hempstead
Frey M, Schullehner K, Dick R et al (2009) Benzoxazinoid biosynthesis, a model for evolution of secondary metabolic pathways in plants. Phytochemistry 70:1645–1651
Fukuoka S, Saka N, Koga H et al (2009) Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325:998–1001
Gallego F, Calles B, Benito C (1998) Molecular markers linked to the aluminium tolerance gene Alt1 in rye (Secale cereale L.) Theor Appl Genet 97:1104–1109
Groszyk J et al (2013) Adaptation of VIGS system to functional analysis of genes controlling the biosynthesis of DIBOA in rye. 4th Polish Congress of Genetics 10th–13th September, Poznań, Poland
Groszyk J et al (2015) International Conference on Rye Breeding and Genetics, 24th–26th June, Wrocław, Poland
Haseneyer G, Schmutzer T, Seidel M et al (2011) From RNA-seq to large-scale genotyping—genomics resources for rye (Secale cereale L.) BMC Plant Biol 11:31
Izumi K, Yamaguchi I, Wada A et al (1984) Effects of a new plant-growth retardant (E)-1-(4-chlorophenyl)-4,4-dimethyl-2-(1,2,4-triazol-1-Yl)-1-penten-3-Ol (S-3307) on the growth and gibberellin content of rice plants. Plant Cell Physiol 25:611–617
Kaneko M, Itoh H, Ueguchi-Tanaka M et al (2002) The alpha-amylase induction in endosperm during rice seed germination is caused by gibberellin synthesized in epithelium. Plant Physiol 128(4):1264–1270
Kato-Noguchi H (2008) Effects of four benzoxazinoids on gibberellins induced α-amylase activity in barley seeds. J Plant Physiol 165:1889–1894
Kertho A, Mamidi S, Bonman JM et al (2015) Genome-wide association mapping for resistance to leaf and stripe rust in winter-habit hexaploid wheat landraces. PLoS One 10(6):e0129580
Khlestkina EK, Than MHM, Pestsova EG, Röder MS, Malyshev SV et al (2004) Mapping of 99 new microsatellite-derived loci in rye (Secale cereale L.) including 39 expressed sequence tags. Theor Appl Genet 109:725–732
Korzun V, Malyshev S, Voylokov AV, Börner A (2001) A genetic map of rye (Secale cereale L.) combining RFLP, isozyme, protein, microsatellite and gene loci. Theor Appl Genet 102:709–717
Krattinger SG, Lagudah EXS, Spielmeyer W et al (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323(5919):1360–1363
La Hovary C (2012) Allelochemicals in Secale cereale: biosynthesis and molecular biology of benzoxazinones. Ph.D. dissertation. North Carolina State University http://gradworks.umi.com/34/63/3463787.html
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Li Y, Böck A, Haseneyer G, Korzun V et al (2011) Association analysis of frost tolerance in rye using candidate genes and phenotypic data from controlled, semi-controlled, and field phenotyping platforms. BMC Plant Biol 11:146
Li H, Vikram P, Singh RP et al (2015) A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genomics 16(1):216
Liu S, Wang X, Wang H et al (2013) Genome-wide analysis of ZmDREB genes and their association with natural variation in drought tolerance at seedling stage of Zea mays L. PLoS Genet 9(9):e1003790
Liu Z, Zhu C, Jiang Y, Tian Y, Yu J, An H, Tang W, Sun J, Tang J, Chen G, Zhai H, Wang C, Wan J (2016) Association mapping and genetic dissection of nitrogen use efficiency-related traits in rice (Oryza sativa L.) Funct Integr Genomics 16(3):323–333
Lohwasser U, Rehman Arif MA, Börner A (2013) Discovery of loci determining pre-harvest sprouting and dormancy in wheat and barley applying segregation and association mapping. Biol Plant 57(4):663–674
Makowska B, Bakera B, Rakoczy-Trojanowska M (2015) The genetic background of benzoxazinoid biosynthesis in cereals. Acta Physiol Plant 31:176
Malosetti M, Ribaut J-M, van Eeuwijk FA (2013) The statistical analysis of multi-environment data: modeling genotype-by-environment interaction and its genetic basis. Front Physiol 4:44
Mares DJ, Mrva K (2014) Wheat grain preharvest sprouting and late maturity alpha-amylase. Planta 240(6):1167–1178
Martis MM, Zhou R, Haseneyer G, Schmutzer T, Vrána J, Kubaláková M, König S, Kugler KG, Scholz U, Hackauf B, Korzun V, Schön CC, Dolezel J, Bauer E, Mayer KFX, Stein N (2013) Reticulate evolution of the rye genome. Plant Cell 25:3685–3698
Masojć P, Larsson-Raźnikiewicz M (1991) Genetic variation of α-amylase levels among rye (Secale cereale L.) kernels, tested by gel diffusion technique. Swedish J Agric Res 21:141–145
Masojć P, Milczarski P (2009) Relationship between QTL for preharvest sprouting and alpha-amylase activity in rye grain. Mol Breeding 23:75–84
Masojć P, Myśków B, Milczarski P (2001) Extending a RFLP-based genetic map of rye using random amplified polymorphic DNA (RAPD) and isozyme markers. Theor Appl Genet 102:1273–1279
Masojć P, Lebiecka K, Milczarski P et al (2009) Three classes of loci controlling preharvest sprouting in rye (Secale cereale L.) discerned by means of bidirectional selective genotyping (BSG). Euphytica 170:123–129
Masojć P, Wiśniewska M, Łań A et al (2011) Genomic architecture of alpha-amylase activity in mature rye grain relative to that of preharvest sprouting. J Appl Genet 52(2):153–160
Miedaner T, Sperling U (1995) Effect of brown rust on yield components of winter rye hybrids and assesment of quantitative resistance. J Phytopathol 143:725–730
Miedaner T, Gey A, Sperling U, Gieger H (2002) Quantitative-genetic analysis of leaf-rust resistance in seedling and adult-plant stages of inbred lines and their testcrosses in winter rye. Plant Breed 121:475–479
Miedaner T, Klocke B, Flath K et al (2012) Diversity, spatial variation, and temporal dynamics of virulences in the German brown rust (Puccinia recondite f sp secalis) population in winter rye. Eur J Plant Pathol 132:23–35
Milczarski P, Banek-Tabor A, Lebiecka K et al (2007) New genetic map of rye composed of PCR-based molecular markers and its alignment with the reference map of the DS2 × RXL10 intercross. J Appl Genet 48(1):11–24
Milczarski P, Bolibok-Brągoszewska H, Myśków B et al (2011) A high density consensus map of rye (Secale cereale L.) based on DArT markers. PLoS One 6(12):e28495
Milczarski P, Hanek M, Tyrka M, Stojałowski S (2016) The application of GBS markers for extending the dense genetic map of rye (Secale cereale L.) and the localization of the Rfc1 gene restoring male fertility in plants with the C source of sterility-inducing cytoplasm. J Appl Genet 57(4):439–451
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acid Res 8:4321–4325
Myśków B, Stojałowski S, Łań A et al (2012) Detection of the quantitative trait loci for α-amylase activity on a high-density genetic map of rye and comparison of their localization to loci controlling preharvest sprouting and earliness. Mol Breeding 30(1):367–376
Niemeyer HM (2009) Hydroxamic acids derived from 2-hydroxy-2H-1,4-benzoxazin-3(4H)-one: key defense chemicals of cereals. J Agr Food Chem 57:1677–1696
Philomin J, Rutkoski JE, Poland JA et al (2015) Genome -wide association mapping for leaf tip necrosis and pseudo-black chaff in relation to durable rust resistance in wheat. The Plant Genome 8(2):1-12
Rafalski JA (2010) Association genetics in crop improvement. Curr Opin Plant Biol 13:174–180
Rakoczy-Trojanowska M, Orczyk W, Krajewski P, Bocianowski J, Stochmal A, Kowalczy M (2016) ScBx gene based association analysis of hydroxamate content in rye (Secale cereale L.) J Appl Genet 56(3):287–298
Roux SR, Wehling P (2010) Nature of mixed infection type 2(5) observed in rye (Secale cereale L.) plants carrying the Pr1 leaf-rust resistance gene. J Kulturpflanzen 62:29–34
Roux SR, Hackauf B, Linz A et al (2004) Leaf-rust resistance in rye (Secale cereale L.). 2. Genetic analysis and mapping of resistance genes Pr3, Pr4 and Pr5. Theor Appl Genet 110:192–201
Saal B, Wricke G (1999) Development of simple sequence repeat markers in rye (Secale cereale L.) Genome 42:964–972
Sansaloni C, Petroli C, Jaccoud D et al (2011) Diversity arrays technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc 5(Suppl 7):54
Saxena RK, Penmetsa RV, Upadhyaya HD et al (2012) Large-scale development of cost-effective single-nucleotide polymorphism marker assays for genetic mapping in pigeonpea and comparative mapping in legumes. DNA Res 19(6):449–461
Semagn K, Babu R, Hearne S, Olsen M (2014) Single nucleotide polymorphism genotyping using Kompetitive allele specific PCR (KASP): overview of the technology and its application in crop improvement. Mol Breeding 33:1–14
Solodukhina OV (1997) Genetics and identification of rust resistance in rye. J Appl Genet 38B:111–116
Solodukhina OV (2002) Genetic characterization of rye accessions with regard to brown rust resistance. Russ J Genet 38:399–407
Sue M, Nakamura C, Nomura T (2011) Dispersed benzoxazinone gene cluster: molecular characterization and chromosomal localization of glucosyltransferase and glucosidase genes in wheat and rye. Plant Physiol 157:985–997
Twardowska M, Masojć P, Milczarski P (2005) Pyramiding genes affecting sprouting resistance rye by means of marker assisted selection. Euphytica 143:257–260
Wehling P, Linz A, Hackauf B et al (2003) Leaf-rust resistance in rye (Secale cereale L.). 1. Genetic analysis and mapping of resistance genes Pr1 and Pr2. Theor Appl Genet 107(3):432–438
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178(1):539–551
Zhu C, Gore M, Buckler EXS, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgments
This work was financially supported by the National Centre for Research and Development, Project No. PBS1/A8/12/2012.
Author information
Authors and Affiliations
Contributions
MRT—conceived and designed the experiments and wrote the manuscript. WO—helped in research design. PK and JB—analysed the data. MS, WW, KB—organized and conducted the experiments on brown rust. Pawel M, Piotr M, KB, ZB, WB—organized and conducted experiments on pre-harvest sprouting and alpha-amylase. Pawel M and Piotr M—mapped DArT-SNPs. MTK—conducted experiments on evaluation of associations.
All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 549 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rakoczy-Trojanowska, M., Krajewski, P., Bocianowski, J. et al. Identification of Single Nucleotide Polymorphisms Associated with Brown Rust Resistance, α-Amylase Activity and Pre-harvest Sprouting in Rye (Secale cereale L.). Plant Mol Biol Rep 35, 366–378 (2017). https://doi.org/10.1007/s11105-017-1030-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-017-1030-6