
Abstract
Quantitative genetic studies in natural populations are of growing interest to speciation research since divergence is often believed to arise through micro-evolutionary change, caused by natural selection on functional morphological traits. The species flock of cichlid fishes in Africa’s oldest lake, Lake Tanganyika, offers a rare opportunity to study this process. Using the cichlid species Tropheus moorii, we assessed the potential for microevolution in a set of morphological traits by estimating their quantitative genetic basis of variation. Two approaches were employed: (1) estimation of trait heritabilities (h 2) in situ from a sample of wild caught fish, and (2) estimation of h 2 from first generation offspring produced in a semi-natural breeding experiment. In both cases, microsatellite data were used to infer pedigree structure among the sampled individuals and estimates of h 2 were made using an animal model approach. Although power was limited by the pedigree structures estimated (particularly in the wild caught sample), we nonetheless demonstrate the presence of significant additive genetic variance for aspects of morphology that, in the cichlid species Tropheus moorii, are expected to be functionally and ecologically important, and therefore likely targets of natural selection. We hypothesize that traits showing significant additive genetic variance, such as the mouth position have most likely played a key role in the adaptive evolution of the cichlid fish Tropheus moorii.
Similar content being viewed by others

Introduction
Since the discovery of the cichlid fish species flocks in the East African Great Lakes Victoria, Malawi, and Tanganyika (Boulenger, 1898), their unsurpassed capacity for rapid diversification has fascinated evolutionary biologists. Greenwood (1984) pointed to the correlation between the age of a radiation and the average morphological divergence among lake endemics. Thus, in Lake Victoria which is at most 200,000 years old, there are no extreme morphotypes and divergent species are “connected” by phenotypically intermediate species in morphoclines (Seehausen, 2002, 2004; Verheyen et al., 2003). In contrast, the much older species flock in Lake Tanganyika is characterized by far higher average morphological divergence and, in comparison to Victoria, lacks species with intermediate morphotypes. This suggests that morphological diversification is driven by natural selection pushing morphologies toward more diverse and extreme forms (Mayr, 1984). Such selection may result from competition between ecologically and morphologically similar species and in the long run may drive intermediates to extinction as the species flock grows older (Sturmbauer, 1998). Progressive specialization is not restricted to morphological adaptation alone; it is reached by a combination of morphological, behavioral, physiological, and life history traits. The enormous eco-morphological and behavioral diversity of the Lake Tanganyika cichlid species assemblage make it a prime model system to study explosive speciation and adaptive radiation at an advanced stage (Fryer & Iles, 1972; Greenwood, 1984; Meyer, 1993; Sturmbauer, 1998; Kornfield & Smith, 2000; Turner et al., 2001; Kocher, 2004; Salzburger & Meyer, 2004; Seehausen, 2006; Koblmüller et al., 2008; Sturmbauer et al. 2011).
While sexual selection is believed to have played a large role in cichlid diversification, natural selection on functional morphology, in association with niche segregation, is also implicated (Fryer & Iles, 1972; Liem, 1973; Greenwood, 1984; Mayr, 1984). In fact, selection on functional morphological traits associated with feeding and locomotion is believed to have played a major role in the diversification or adaptive radiation of several freshwater fish taxa, including arctic charr, whitefish, and sticklebacks as well as cichlid fishes (Liem, 1973; Bernatchez et al., 1999; Schluter, 2000; Albertson et al., 2003; McKinnon et al., 2004; Salzburger, 2009; Hudson et al., 2011). While it is, therefore, likely that morphological traits have been, and continue to be, under natural selection, any evolutionary response to selection is contingent on the presence of additive genetic variation underlying observed phenotypic variation. Currently, data are rapidly accumulating on the genetic architecture of those morphological traits expected to be of importance to cichlid diversification (Albertson et al., 2003; Albertson & Kocher, 2006; Streelman & Albertson, 2006; Streelman et al., 2007; Loh et al., 2008; Salzburger, 2009).
While quantitative genetic data are increasingly used to estimate genetic parameters in situ in wild vertebrate populations (e.g. Kruuk et al., 2000; Kruuk, 2004; Coltman, 2005; Ellegren & Sheldon, 2008), studies of fishes are scarce (Wilson et al., 2003a; Garant et al., 2003). This deficiency is in large part due to the requirement for pedigree structure to be present (and identifiable) within a sample of individuals. This requirement can be particularly difficult to meet in many natural fish species, particularly if effective population sizes are high, and spatial and temporal overlap of relatives is low (Wilson & Ferguson, 2002). Conversely, limited dispersal will increase the probability of sampling related individuals from a wild population (Wilson & Ferguson, 2002) and for this reason we chose the philopatric cichlid fish species Tropheus moorii (Baric et al., 2003; Sturmbauer et al., 2005; Sefc et al., 2007) for the present study, the primary goal of which was to estimate genetic variance for functional morphological traits, into assess the potential for microevolutionary responses to selection acting on them.
Tropheus moorii is strictly adapted to live in rock- or cobble shores and shows high levels of within-species diversity with about 120 distinctly colored “geographical races” having been described (Konings, 1998, Schupke, 2003). Behavioral studies in the field have shown that the species lives in a complex social system (Yanagisawa & Nishida, 1991; Sturmbauer & Dallinger, 1995; Egger et al., 2006; Sefc, 2008). Here, we selected a wild island population which is enclosed by dispersal barriers in the southern basin of the lake. We used pedigree information and phenotypic data to estimate quantitative genetic parameters following two approaches. Firstly, we attempted to estimate parameters in situ by reconstructing pedigree relationships among a wild caught sample of adult fish. Secondly, because the effective population size of T. moorii is known to be high (Sefc et al., 2007) and the success of obtaining a suitable sample composition (with respect to relationship structure) could not be known a priori, we also used wild caught parents to produce an offspring generation in a semi-natural pond breeding experiment.
Materials and methods
Sampling
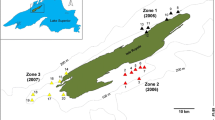
On the basis of recent work estimating parent–offspring relationships in wild Tropheus populations, we selected a population from Mbita Island in the very South of Lake Tanganyika, which is likely to yield a sample with sufficient pedigree structure to permit quantitative genetic analyses (Fig. 1; for details see Koch et al., 2008). The present study is based upon a total of 365 specimens contained within two overlapping data sets, subsequently denoted as “Wild population” (n = 241) and “Pond population” (n = 224). The wild population sample (n = 241: 56 males, 133 females, and 52 juvenile) was collected via net-catching by divers in March 2005 at Mbita Island [S 08°44′; E 31°06′] in Zambia. 141 specimens (31 males, 58 females, and 52 juvenile) were shipped to Austria alive in March 2005. Digital images of the individuals for geometric morphometric analysis were obtained using a flatbed scanner (for details on scanning method please refer to Herler et al., 2007), fish were fin-clipped and sexed via dissection. The remaining 100 wild caught fish (25 males and 75 females) were kept in a concrete pond (1.5 m × 3.5 m, 70 cm deep) situated at Lake Tanganyika and served as the parental generation in our breeding experiment. These adults were allowed to spawn freely and, together with their offspring, comprise the Pond data set. The parental fish were removed from the pond after 1 year and shipped to Austria alive, scanned for geometric morphometric analysis, fin-clipped and sexed. The F1 generation (n = 124 juvenile) were anesthetized, fin-clipped and scanned in March 2007, and then returned to the pond.

Sampling site of the investigated island population of Tropheus moorii at Mbita Island in the southern basin of Lake Tanganyika [S 08°44′; E 31°06′]
Phenotypic measurements
Morphological traits were defined and measured for all specimens using both geometric and traditional morphometric approaches (Bookstein, 1991; Rohlf, 1999; Slice, 2001; Sheets, 2003). Firstly, body shape was quantified via a multivariate statistic. Residuals of the relative warp (RW) and canonical variate (CV) analysis were used to describe phenotypic differences (Maderbacher et al., 2008; Herler et al., 2010). Note that this is a geometric morphometric method that captures differences in overall body shape based upon 19 landmarks (Maderbacher et al., 2008; Fig. 2). Secondly, traditional morphometric measurements in the form of inter-landmark distances (ILD; revered as truss nets in Bookstein, 1991) were used as morphological variables (for details see Maderbacher et al., 2008), resulting in 19 discrete morphological measurements revered as ILD 1–19 (Fig. 2). For each specimen, ILD estimates were expressed as proportions of standard length (from the tip of the lower jaw to the posterior end of the hypural bone) in order to reduce allometric effects.

Landmark positions (LM 1–19; shown in white numerals) and inter-landmark distances (ILD 1–19; shown in black numerals) utilized in geometric morphometrics (for details see also Maderbacher et al., 2008)
Pedigree reconstruction
Pedigree information was estimated from microsatellite data as described in Koch et al. (2008), with exact procedures differing between data sets. Firstly, in the wild sample we used a sibship reconstruction approach implemented in the software program COLONY (Wang, 2004) using nine microsatellite loci to identify pedigree structure. This method assumes that individuals are either full sibs or unrelated. Clearly, this is a simple and likely erroneous model of the true pedigree. However, errors in the reconstructed pedigree structure are expected to cause downward bias such that estimates of additive genetic (co)variances will be conservative (Wilson et al., 2003b; Kruuk, 2004). In the Pond sample, which was known a priori to contain parental and offspring individuals, we performed parentage assignment using genotypic data for 10 microsatellite loci. We estimated the Pond pedigree using the software program PAPA (Duchesne et al., 2002) under two assumed genotypic error rates (pedigree 1 with an assumed error rate of 0, and pedigree 2 with an assumed error rate of 1%).
Quantitative genetic analysis
The morphological traits studied herein build on two previous studies, in which a landmark-based geometric morphometric characterization of various Tropheus populations was elaborated (Maderbacher et al., 2008; Herler et al., 2010). Technical details about the generation of the underlying phenotypic trait estimates (landmarks, trait loadings, and deformation grids, explained variances for multivariate population comparisons) can be obtained therein. Additive genetic variance components were estimated for phenotypic traits using the reconstructed pedigrees in an animal model approach (Henderson, 1984; Lynch & Walsh, 1998; Kruuk, 2004). The animal model is a particular case of a linear mixed effect model specified as:
where y is a vector of the studied phenotypic values, b is a vector of fixed effects, u is a vector of random effects that includes the additive genetic effect. X and Z are design matrixes that relate the appropriate fixed and random effects to each individual’s phenotype and e is a vector of residual errors. In addition to the mean, fixed effects of sex, and origin were fitted as two-level factors. The former was included to account for known sexual dimorphism in body shape (Herler et al., 2010), while the latter was used in analyses of the Pond data set to account for the fact that the parental fish were wild caught and the F1 were pond-bred. Note origin is, therefore, also a surrogate for generation that should also account for any phenotypic differences arising from age or specimen treatment (parental fish were phenotyped after death and formalin preserved). Phenotypic variance was then partitioned into additive genetic variance V A and residual (environmental) variance (V R). The significance of the additive genetic variance was assessed for each trait using likelihood ratio tests (i.e., comparison to a reduced model with no additive effect fitted), and heritability (h²) was estimated as the proportion of additive genetic variance (V A) to total phenotypic variance (V P). V P was estimated as the sum of V A and V R and h 2 should, therefore, be interpreted as the proportion of phenotypic variance remaining after conditioning on the fixed effects that is explained by additive effects (Wilson, 2008). We also estimated the coefficient of additive genetic variation CVA to provide a less scale sensitive measure for comparing additive genetic variance across populations and environments Houle (1992). CVA was calculated as 100 × \( \sqrt {V_{\text{A}} } \)/\( \bar{x} \) (where \( \bar{x} \) is the trait mean). We also attempted to fit bivariate animal models in order to estimate genetic correlations (r G) among morphometric traits. However, in both data sets (but especially in the Wild population data) statistical uncertainty was too great to allow any meaningful biological interpretation of the results, while convergence problems were encountered for many pairs of traits. We believe this to reflect insufficient pedigree data to properly support parameterization of these more complex models and consequently we do not present these models (but see later discussion on this limitation of the current study). All models were implemented using the program ASReml (Gilmour et al., 2002) and significance of additive genetic variance was assed via likelihood ratio test statistics (LRT).
Results
Wild population data set
Pedigree
Pedigree reconstruction using COLONY (Wang, 2004) grouped the 241 specimens into 133 full-sib families. Family sizes ranged from 1 (n = 30) to 3 (n = 5) specimens, with the majority of families containing just two individuals (n = 98; data not shown). Given the strong assumptions of this sib-ship partitioning (i.e., individuals in the sample are either full sibs or unrelated), we fully acknowledge that this pedigree structure will certainly contain many errors. However, under the univariate animal model fitted these pedigree errors should induce downward bias and thus result in conservative heritability estimates (Morrissey et al., 2007).
Quantitative genetic analyses
Estimates of heritability ranged from 0 to 0.72 among the measured morphometric traits with a median estimate of 0.16. In general, standard errors were large indicating a lack of precision that is expected given the limited amount of (estimated) relationship structure in the wild sample. Note that for several traits, genetic variance was constrained to the boundary of biologically permissible parameter space (i.e. negative values of V A are not generally interpretable) such estimated h 2 also equals zero. For these traits, the uncertainty around h 2 cannot be estimated and no standard error is presented in Table 1. Nonetheless, estimates of additive genetic variance (V A) were statistically significant for 5 out of the 24 morphological traits (Table 1). These traits included truss lengths (i.e., inter-landmark distances (ILD)), ILD3 (h 2 0.67; s.e. 0.25), ILD5 (h 2 0.61; s.e. 0.24), ILD10 (h 2 0.41; s.e. 0.24) and ILD13 (h 2 0.72; s.e. 0.25). However, V A for body shape, as estimated in the form of multivariate residuals, was only significant for one canonical variate (CV1: h 2 0.54; s.e. 0.26; Table 1).
Pond population data set
Pedigree
Assignment of parental pairs with PAPA (Duchesne et al., 2002) used genotypic data at 10 microsatellite loci for 124 offspring sampled from the pond, and 88 of the adults initially stocked treated as potential parents. Under an assumption of zero genotyping error (pedigree 1), parental pairs were assigned to 62% of the offspring (i.e. 71 individuals in total), with no assignment possible for 37%, and 0.8% (i.e. one individual) having ambiguous assignment (i.e. more than one parental pair getting the highest likelihood score).
With an assumed genotyping error rate of 1%, 99% of the offspring (i.e. 123 individuals) were successfully assigned to a single parental pair with the single ambiguous assignment remaining. Note that trait heritabilities were estimated using both pedigrees since the comparison in and of itself is potentially interesting. Although we cannot know the true pedigree, it is not unreasonable to assume that all assigned parental pairs in Pedigree 1 are correct, but since not all parentage was assigned, errors are incorporated in the form of true parent-offspring pairs that are assumed to be unrelated. Conversely, by allowing for genotyping error Pedigree 2 assigned parentage to all but one offspring, reducing the amount of unrecognized relationship structure but with a higher risk of making incorrect assignments.
Quantitative genetic analyses
Using Pedigree 1 to parameterize our animal models, estimates of heritability ranged from 0 to 0.96 with a median of 0.34. There was evidence of additive genetic variance in 10 of the 24 traits tested. Note that for the single trait of ILD11DFB, we were unable to obtain a stable model convergence and consequently no estimate of h 2 is presented. In detail, using Pedigree 1 we estimated significant additive genetic variance for: inter-landmark distance 1 (ILD1: h 2 0.96; s.e. 0.20), ILD4 (h 2 0.45; s.e. 0.20), ILD5 (h 2 0.96; s.e. 0.16), ILD9SNL (h 2 0.56; s.e. 0.25), ILD13 (h 2 0.78; s.e. 0.23), ILD14 (h 2 0.75; s.e. 0.20), ILD15CPL (h 2 0.48; s.e. 0.20), ILD18 (h 2 0.65; s.e. 0.23) and in terms of body shape for: relative warp score 1 (RW1: h 2 0.84; s.e. 0.20) and RW3 (h 2 0.53; s.e. 0.22; for details see Table 1).
Under Pedigree 2, h 2 estimates ranged from 0.16 to 0.71 with a median of 0.24, and additive genetic variance was significant for 16 of the traits tested. In detail, significant results were obtained for ILD1 (h 2 0.61; s.e. 0.18), ILD2 (h 2 0.28; s.e. 0.18), ILD4 (h 2 0.36; s.e. 0.17), ILD5 (h 2 0.42; s.e. 0.19), ILD7 (h 2 0.18; s.e. 0.15), ILD8HL (h 2 0.39; s.e. 0.18), ILD9SNL (h 2 0.36; s.e. 0.19), ILD11DFB (h 2 0.65; s.e. 0.21), ILD14 (h 2 0.71; s.e. 0.17), ILD15CPL (h 2 0.40; s.e. 0.18), ILD16 (h 2 0.24; s.e. 0.16), ILD18 (h 2 0.26; s.e. 0.18), ILD19ED (h 2 0.24; s.e. 0.18) and in terms of body shape for relative warp score 2 (RW2: h 2 0.22; s.e. 0.17), RW3 (h 2 0.41; s.e. 0.19) and for canonical variate 2 (CV2: h 2 0.16; s.e. 0.15; for details see Table 1).
Comparing the estimates from the two estimated Pond, pedigree structures suggests a significant difference (Pairwise Wilcoxon matched pairs test, V = 236.5, P = 0.002) with, on average, higher values obtained using Pedigree 1. The standard errors of the h2 estimates also differ (Pairwise Wilcoxon matched pairs test, V = 182, P < 0.001), and are slightly smaller under Pedigree 2. This pattern is consistent with the expectation that Pedigree 2 affords greater precision due to the higher number of relationships assigned, but perhaps at a cost of increased downward bias arising from erroneous assignments.
Discussion
Quantitative genetic studies in free ranging natural populations are of particular interest for understanding (micro-evolutionary) changes in phenotypic traits. This study addressed trait evolution using a Lake Tanganyika model species for allopatric divergence, Tropheus moorii. We addressed the potential of particular morphometric traits to respond to different selective forces by taking two approaches: firstly by estimating genetic variance in situ from wild caught individuals, and secondly by using a pond-bred population maintained and allowed to breed under semi-natural conditions. By taking this two-fold approach, heritable genetic components were indeed demonstrated for several quantitative traits in both data sets.
By studying a natural population of the species Tropheus moorii, in which the degree of relatedness was expected to be high due to philopatry (Koch et al., 2008), we were able to detect significant additive genetic variance (V A) and heritability (h 2) for five quantitative traits including body shape (see Table 1). With respect to the understanding the pathways of ecological niche differentiation, our finding of significant heritability for the mouth position (ILD 10) is perhaps most interesting. Indirectly, this suggests a genetic basis for the mode of food uptake, as mouth shape and position is reflected in the biting angle of epilithic algae feeders (see also Albertson et al., 2005). It has been hypothesized that the great potential of cichlid fishes to respond to novel trophic niches lies in their capacity for rapid evolution allowing exploitation of new food resources (Liem, 1973). The potential for adaptive evolutionary response might also be substantiated for other quantitative traits of ecological significance showing significant V A and h 2 values: head length, snout length, and eye diameter. Moreover, fin length might be a measure of swimming ability. As many of these structures are connected to adaptation to particular dietary specializations, this is in line with the key innovation hypothesis of Liem (1973) for the evolutionary success of cichlid fishes in general. This study is the first to demonstrate significant additive genetic variance in specific traits, and hence the potential for trait evolution in a natural cichlid fish population.
In situ estimation of quantitative genetic parameters is desirable in the sense that it allows us to estimate levels of genetic variance that are actually expressed under natural conditions (i.e., the context in which selection operates). However, the challenges of obtaining a sample with a relationship structure suitable for quantitative genetic analyses can be considerable for fish systems (Wilson & Ferguson, 2002), and we fully acknowledge that our heritability estimates here are based on small amounts of likely quite inaccurate pedigree data. Consequently, the lack of precision as reflected in the estimated standard errors, is not unexpected. Future studies could likely (at least) double the sample size without affecting the species community of continuous rock dwelling habitats (although biological conservation might have to be considered for some small cichlid fish populations). However, we emphasize that it is the ability to sample (and identify) relatives, rather than the sample size alone, that limits the ability to make robust inferences about the genetic basis of trait inheritance.
Conversely, while the genetic parameters estimated from pond-reared fish may be less representative of those in the wild population (Roff, 1997, 2002), this approach does ensure that a sample will contain more relatedness structure, and (since parents and offspring are know to be sampled, almost completely) also provides a far more tractable problem for the pedigree estimation from molecular data. Thereby we detected significant heritability for several morphological traits relevant for ecological specialization, e.g. head length, snout length, eye diameter, standard length, and length of the caudal peduncle (Table 1). Interestingly, h 2 and CVA estimates are somewhat suggestive of lower levels of genetic variance for inter-landmark distances associated with head morphology (as opposed to those associated with the whole body). Although we note that this is a post-hoc observation unsupported by statistical analyses, if robust it could be viewed as consistent with erosion of within-population genetic variance for head morphology by strong selection in the past, with a corresponding increase in the relative contribution of plasticity to observed (within-population) variance. Note that such a scenario is perfectly consistent with finding that genetic variation makes a major contribution to among-lineage divergence in trophic morphology (Albertson & Kocher, 2006).
Our use of alternative estimated pedigree structures for the pond fish also yielded results consistent with the expectation that pedigree error will systematically reduce heritability estimates (Thomas et al., 2002), at least under a simple model such as that used herein (see Morrissey et al., 2007 for discussion). This is indicated by the reduction of additive genetic variance in several quantitative traits when a less stringent pedigree accepting more false assignments was used as the underlying pedigree (e. g. ILD4, ILD5, ILD7, ILD9SNL, ILD14, ILD15CPL, ILD16, ILD18, ILD19ED, and ILD1SL; see also Table 1). Our intension was to quantify within-population genetic variance for ecologically meaningful phenotypic traits rather than detecting genetic variation contributing to among population differences.
While we were able to infer the presence of additive genetic variance for morphometric traits from two generations of pond data, more stringent analyses including F2 and F3 are planned although data are not yet available due to the relatively long generation time (approximately 2.5 years). Data from additional generations will not only improve statistical power for estimating heritabilities (and allow extension to multivariate analyses) but it will also facilitate more effective investigations of additional sources of phenotypic variance, including maternal effects (which if present may confound heritability estimates), and phenotypic plasticity (which has been shown to change the phenotype significantly in this species within one generation in a standardized pond environment; Kerschbaumer et al., 2011).
Conclusions
By utilizing the animal model approach, we could, for the first time, demonstrate significant additive genetic effects for certain quantitative traits in a wild cichlid fish population of Lake Tanganyika. Furthermore, the results from the breeding design imply evolutionarily significant heritabilities for some quantitative traits, and we suggest that these traits should be further investigated in character displacement experiments, to isolate those characters playing the key role in adaptive evolution of the genus Tropheus. Ecological character displacement experiments can be designed in the future involving pond breeding settings which will facilitate further quantitative genetic analyses of trait (co)variation and its genetic component. Also, replicate sampling of other wild populations should be carried out.
References
Albertson, R. C. & T. D. Kocher, 2006. Genetic and developmental basis of cichlid trophic diversity. Heredity 97: 211–221.
Albertson, R. C., J. T. Streelman & T. D. Kocher, 2003. Directional selection has shaped the oral jaws of Lake Malawi cichlid fishes. Proceedings of the National Academy of Sciences 9: 5252–5257.
Albertson, R. C., J. T. Streelman, T. D. Kocher & P. C. Yelick, 2005. Integration and evolution of the cichlid mandible: Molecular basis of alternate feeding strategies. Proceedings of the National Academy of Sciences USA 102: 16287–16292.
Baric, S., W. Salzburger & C. Sturmbauer, 2003. Phylogeography and evolution of the Tanganyikan cichlid genus Tropheus based upon mitochondrial DNA sequences. Journal of Molecular Evolution 56: 54–68.
Bernatchez, L., A. Chouinard & G. Lu, 1999. Integrating molecular genetics and ecology in studies of adaptive radiation: whitefish, Coregonus sp., as a case study. Biological Journal of the Linnean Society 68: 173–194.
Bookstein, F. L., 1991. Morphometric tools for landmark data: geometry and biology. Cambridge University Press, Cambridge.
Boulenger, G., 1898. Description de deux genres nouveaux de la familie des Salamandridés Bulletin de la Société Zoologique, France, Paris.
Coltman, D. W., 2005. Testing marker-based estimates of heritability in the wild. Molecular Ecolology 14: 2539–2599.
Duchesne, P., G. H. Godbout & L. Bernatechz, 2002. PAPA (Package for the Analysis of Parental Allocation): a computer program for simulated and real parental allocation. Molecular Ecology Notes 2: 191–193.
Egger, B., B. Obermüller, H. Phiri, C. Sturmbauer & K. M. Sefc, 2006. Monogamy in the maternally mouthbrooding Lake Tanganyika cichlid fish Tropheus moorii. Proceedings of the Royal Society London B 273: 1797–1802.
Ellegren, H. & B. C. Sheldon, 2008. Genetic basis of fitness differences in natural populations. Nature 452: 169–175.
Fryer, G. & T. D. Iles, 1972. The Cichlid Fishes of the Great Lakes of Africa: Their Biology and Evolution. TFH Publications, Neptune City, NJ.
Garant, D., J. J. Dodson & L. Bernatchez, 2003. Differential reproductive success and heritability of alternative reproductive tactics in wild Atlantic salmon (Salmo salar L.). Evolution 57: 1133–1141.
Gilmour, A.R., B. J. Gogl, B. R. Cullis, S. J. Welham & R. Thompson, 2002. ASReml User Guide Release 1.0. VSN International Ltd., Heml Hempstead, UK.
Greenwood, P. H., 1984. African cichlids and evolutionary theories. In Echelle, A. A. & I. Kornfield (eds), Evolution of fish species flocks. University of Maine at Orono Press, Orono: 141–154.
Henderson, C.R., 1984 Applications of Linear Models in Animal Breeding. University of Guleph Press, Guleph.
Herler, J., L. Lipej & T. Makovec, 2007. A simple technique for digital imaging of live and preserved small fish specimens. Cybium 31: 39–44.
Herler, J., M. Maderbacher, P. Mitteroecker, L. Postl & C. Sturmbauer, 2010. Sexual dimorphism and population divergence in the Lake Tanganyika cichlid fish Tropheus moorii. Frontiers in Zoology 7: 4.
Houle, D., 1992. Comparing evolvability and variability of quantitative traits. Genetics 130: 195–204.
Hudson, A. G., P. Vonlanthen & O. Seehausen, 2011. Rapid parallel adaptive radiations from a single hybridogenic ancestral population. Proceedings of the Royal Society B 278: 58–66.
Kerschbaumer, M., L. Postl, M. Koch, T. Wiedl & C. Sturmbauer, 2011. Morphological distinctness despite large-scale phenotypic plasticity–analysis of wild and pond-bred juveniles of allopatric populations of Tropheus moorii. Naturwissenschaften 98: 125–134.
Koblmüller, S., K. M. Sefc & C. Sturmbauer, 2008. The Lake Tanganyika cichlid species assemblage: recent advances in molecular phylogenetics. Hydrobiologia 615: 5–20.
Koch, M., J. Hadfield, K. M. Sefc & C. Sturmbauer, 2008. Pedigree estimation in wild cichlid fish populations. Molecular Ecology 17: 4500–4511.
Kocher, T. D., 2004. Adaptive evolution and explosive speciation: the cichlid fish model. National Genetics Reviews 5: 288–298.
Konings, A., 1998. Tanganjika-Cichliden in ihrem natürlichen Lebensraum. Cichlid Press, El Paso: 272.
Kornfield, I. & P. F. Smith, 2000. African cichlid fishes: Model systems for evolutionary biology. Annual Review of Ecological Systematics 31: 163–196.
Kruuk, L. E. B., 2004. Estimating genetic parameters in natural populations using the ‘animal model’. Philosophical Transactions of the Royal Society of London B 359: 873–890.
Kruuk, L. E. B., T. H. Clutton-Brock, J. Slate, J. M. Pemperton, S. Brotherstone & F. E. Guiness, 2000. Heritability of fitness in a wild mammal population. Proceedings of the National Academy of Sciences USA 97: 689–703.
Liem, K. F., 1973. Evolutionary strategies and morphological innovations: cichlid pharyngeal jaws. Systematic Zoology 22: 425–441.
Loh, Y.-H. E., L. S. Katz, M. C. Mims, T. D. Kocher, S. V. Yi & J. T. Streelman, 2008. Comparative analysis reveals signatures of differentiation amid genomic polymorphism in Lake Malawi cichlids. Genome Biology 9: R113.
Lynch, M. & B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, Massachusetts.
Maderbacher, M., C. Bauer, J. Herler, L. Postl, L. Makasa & C. Sturmbauer, 2008. Assessment of traditional versus geometric morphometrics for discriminating populations of Lake Tanganyika cichlid fishes. Journal of Zoological Systematics and Evolutionary Research 46: 153–161.
Mayr, E., 1984. Evolution of fish species flocks: a commentary. In EchelleA, A. & I. Kornfield (eds), Evolution of fish species flocks. University of maine at Orono Press, Orono: 3–11.
McKinnon, J. S., S. Mori, B. K. Blackman, L. David, D. M. Kingsley, L. Jamieson, J. Chou & D. Schluter, 2004. Evidence for ecology’s role in speciation. Nature 429: 294–298.
Meyer, A., 1993. Phylogenetic relationships and evolutionary processes in east African cichlid fishes. Trends in Ecology and Evolution 8: 279–284.
Morrissey, M. B., A. J. Wilson, J. M. Pemberton & M. M. Ferguson, 2007. A framework for power and sensitivity analyses for studies of the quantitative genetics of natural populations, and a case study in Soay sheep (Ovis aries). Journal of Evolutionary Biology 20: 2309–2321.
Roff, D., 1997. Evolutionary Quantitative Genetics. Chapman & Hall, New York, USA.
Roff, D., 2002. Life-History Evolution. Sinauer Associates, Sunderland, MA.
Rohlf, F. J., 1999. Shape statistics: Procrustes superimpositions and tangent spaces. Journal of Classification 16: 197–223.
Salzburger, W., 2009. The interaction of sexually and naturally selected traits in the adaptive radiations of cichlid fishes. Molecular Ecology 18: 169–185.
Salzburger, W. & A. Meyer, 2004. The species flock of East African cichlid fishes: recent advances in molecular phylogenetics and population genetics. Naturwissenschaften 91: 277–290.
Schluter, D., 2000. The Ecology of Adaptive Radiation. Oxford University Press, Oxford, UK: 288.
Schupke, P., 2003. Cichlids of Lake Tanganyika, Part 1: The species of the genus Tropheus. Aqualog, Rodgau, Germany: 190.
Seehausen, O., 2002. Patterns in cichlid fish radiation are compatible with Pleistocene desiccation of Lake Victoria and 14.600 year history for its cichlid species flock. Proceedings of the Royal Society London B 269: 491–497.
Seehausen, O., 2004. Hybridization and adaptive radiation. Trends in Ecology and Evolution 19: 198–207.
Seehausen, O., 2006. African cichlid fish: a model system in adaptive radiation research. Proceedings of the Royal Society London B 273: 1987–1998.
Sefc, K. M., 2008. Variance in reproductive success and the opportunity for selection in a serially monogamous species: simulations of the mating system of Tropheus (Teleostei: Cichlidae). Hydrobiologia 615: 21–35.
Sefc, K. M., S. Baric, W. Salzburger & C. Sturmbauer, 2007. Species specific population structure in rock-specialized sympatric cichlid species in Lake Tanganyika. East Africa. Journal of Molecular Evolution 64: 33–49.
Sheets, H. D., 2003. IMP-Integrated Morphometrics Package. Department of Physics, Canisius College, Buffalo, NY.
Slice, D. E., 2001. Landmark coordinates aligned by Procrustes analysis do not lie in Kendall’s shape space. Systematic Biology 50: 141–149.
Streelman, J. T. & R. C. Albertson, 2006. Evolution of novelty in the cichlid dentition. Journal of Experimental Zoology B 306: 216–226.
Streelman, J. T., R. C. Albertson & T. D. Kocher, 2007. Variation in body size and trophic morphology within and among genetically differentiated populations of the cichlid fish, Metriaclima zebra, from Lake Malawi. Freshwater Biology 52: 525–538.
Sturmbauer, C., 1998. Explosive speciation in cichlid fishes of the African Great Lakes: a dynamic model of adaptive radiation. Journal of Fish Biology 53: 18–36.
Sturmbauer, C. & R. Dallinger, 1995. Diurnal variation of spacing and foraging behavior in Tropheus moorii (Cichlidae) in Lake Tanganyika. Netherlands Journal of Zoology 45: 386–401.
Sturmbauer, C., M. Husemann & P. Danley, 2011. Explosive speciation and adaptive radiation in African cichlid fishes. In Habel J. C. & F. Zachos (eds), Biodiversity Hotspots – Distribution and Protection of Conservation Priority Areas. Springer, Germany.
Sturmbauer, C., S. Koblmüller, K. M. Sefc & N. Duftner, 2005. Phylogeographic history of the genus Tropheus, a lineage of rock-dwelling cichlid fishes endemic to Lake Tanganyika. Hydrobiologia 542: 335–366.
Thomas, S. C., D. W. Coltman & J. M. Pemberton, 2002. The use of marker-based relationship information to estimate the heritability of body weight in a natural population: a cautionary tale. Journal of Evolutionary Biology 15: 92–99.
Turner, G. F., O. Seehausen, M. E. Knight, J. C. Allender & R. L. Robinson, 2001. How many species of cichlid fish are there in African lakes? Molecular Ecology 10: 793–806.
Verheyen, E., W. Salzburger, J. Snoeks & A. Meyer, 2003. Origin of the superflock of cichlid fishes from Lake Victoria, East Africa. Science 300: 325–329.
Wang, J., 2004. Estimating pairwise relatedness from dominant genetic markers. Molecular Ecology 13: 3169–3178.
Wilson, A. J., 2008. Why h2 does not always equal VA/VP. Journal of Evolutionary Biology 21: 647–650.
Wilson, A. J. & M. M. Ferguson, 2002. Molecular pedigree analysis in natural populations of fishes: approaches, applications, and practical considerations. Canadian Journal of Fisheries and Aquatic Sciences 59: 1696–1707.
Wilson, A. J., J. A. Hutchings & M. M. Ferguson, 2003a. Selective and genetic constraints on the evolution of body size in a stream-dwelling salmonid fish. Journal of Evolutionary Biology 16: 584–594.
Wilson, A. J., G. McDonald, H. K. Moghadam, C. M. Herbinger & M. M. Ferguson, 2003b. Marker-assisted estimation of quantitative genetic parameters in rainbow trout, Oncorhynchus mykiss. Genetical Research Cambridge 81: 145–156.
Yanagisawa, Y. & M. Nishida, 1991. The social and mating system of the maternal mouthbrooder Tropheus moorii (Cichlidae) in Lake Tanganyika. Japanese Journal of Ichthyology 38: 271–282.
Acknowledgments
We would like to thank the entire team at the Mpulungu Station of the Ministry of Agriculture and Cooperatives, Republic of Zambia at Lake Tanganyika, and T. Veall, Rift Valley Tropicals Ltd., for assistance during field work and breeding. We are further grateful to two anonymous reviewers for insightful comments and suggestions. This study was financed by the Austrian Science Foundation (FWF grants P17968 and P20994 to CS), as well as by the Commission for Interdisciplinary Ecological Studies of the Austrian Academy of Sciences (Grant 2007-04 to CS). M. Koch and M. Kerschbaumer were funded by the Austrian Science Foundation. AJW was supported by a Natural Environment Research Council (NERC) UK fellowship.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest editors: C. Sturmbauer, C. Albrecht, S. Trajanovski & T. Wilke / Evolution and Biodiversity in Ancient Lakes
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Koch, M., Wilson, A.J., Kerschbaumer, M. et al. Additive genetic variance of quantitative traits in natural and pond-bred populations of the Lake Tanganyika cichlid Tropheus moorii . Hydrobiologia 682, 131–141 (2012). https://doi.org/10.1007/s10750-011-0785-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-011-0785-2