Introduction
Hepatitis C virus (HCV) is a positive-strand enveloped RNA virus of the family Flaviviridae, genus Hepacivirus and is one of the commonest causes of chronic hepatitis, cirrhosis and hepatocellular carcinoma in the Western World. However, the recent development and clinical implementation of effective curative treatment strategies using direct acting agents (DAAs) in high income countries has been a welcome success story and is already beginning to turn the tide leading to visions of elimination in the foreseeable future.
Viral discovery
At first the discovery of HCV was frustrating. Identification of two distinct diseases during the 1960s and 1970s, infectious hepatitis (Hepatitis A) and serum hepatitis (Hepatitis B), resulted in diagnostic developments, which in turn helped establish the existence of an additional non-A non-B hepatitis (NANBH) of unknown aetiology (Ref. Reference Pawlotsky1). In 1989, immunoscreening of an expression library with post-transfusion NANBH serum led to the identification of an enveloped positive-stranded RNA molecule, with a length of at least 10 000 nucleotides, related to the togaviridae or flaviviridae (Ref. Reference Choo2) and subsequently classified into the Hepacivirus genus within the Flaviviridae family. In this regard, discovery of HCV was atypical and marked a new direction in the history of virology, in that it relied upon molecular approaches rather than classical methods such as culturing the virus and imaging of viral particular under electron microscopy.
Epidemiology
In 2015, the World Health Organization (WHO) provided a series of telling statistics. They estimated that 71 million individuals, 1% of the World's population, were living with HCV (Ref. 3). Of those, only 14 million had a formal diagnosis and just 1.1 million had started treatment. Whilst the cumulative number of people treated for HCV had risen to 5.5 million in 2015, only 0.5 million of these had received DAAs. Encouragingly, even prior to the widespread availability of DAAs, estimates of the total number of individuals with a successful treatment outcome exceeded estimates of those dying from end-stage HCV infection (843 000 and 399 000, respectively), although the highest rates of treatment success were linked to those regions with access to new DAAs.
On a more cautious note in the same report, there were more newly acquired HCV infections (1.75 million) than patients beginning treatment of any kind. The leading causes of acquisition were unsafe healthcare practices and intravenous drug use (23% of cases). An estimated 8% of infected individuals, 5.6 million people, continue to inject drugs and of the 36.7 million individuals living with HIV in 2015, an estimated 2.3 million were also infected with HCV.
Whilst hepatitis C is a global disease, the distribution is uneven; in general, European and Eastern Mediterranean regions are those most affected, with a reported prevalence of 1.5 and 2.3%, respectively. The leading cause of transmission in Europe is injecting drug use, whilst transmission in the Eastern Mediterranean is more often associated with unsafe healthcare.
Thus, it is unsurprising that rates of diagnosis are higher in higher income countries. First-line testing is available for US$0.5–3, although viral genotyping, used to guide drug selection, is much more costly, adding between US$25 and US$200 to diagnostic costs. Generic DAAs can now be accessed for as little as US$200 for a treatment course in some countries, although prices can reach as high as US$45 000.
The recognition of HCV as an on-going global burden, alongside the availability of highly effective treatments led to the WHO adoption of the first ‘Global Health Sector Strategy on Viral Hepatitis 2016–2021’. This aims to maximise the impact on viral hepatitis by targeting a reduction in new infections by 90% and reducing deaths by 65% by 2030.
Natural course of infection
Acute infection
Occurring 2 weeks to 6 months following acquisition, acute infection is rarely symptomatic and so classically remains unrecognised. As such, appropriate tests are seldom undertaken. Symptomatic patients usually exhibit findings common to many acute viral infections – fatigue, anorexia, nausea, low-grade fever and mild abdominal pain, whilst only a minority present with a syndrome of viral hepatitis, with the development of jaundice seen in <25%.
Retrospective studies of post-transfusion hepatitis indicate that between 18 and 34% of individuals with acute infection clear HCV (Ref. Reference Westbrook and Dusheiko4). Symptomatic patients are somewhat more likely to clear HCV and studies evaluating time to clearance have been confined mostly to this group. Grebely et al. reviewed nine prospective international cohorts (the majority of which comprised people who injected drugs, PWID) revealing spontaneous clearance in 173 of 632 participants. Median time to clearance was 16.5 weeks, with 34, 67 and 83% of those demonstrating clearance at 3, 6 and 12 months, respectively (Ref. Reference Grebely5). Female sex was the strongest predictor of clearance, but host genetic polymorphisms and HCV genotype 1 infection were also associated with increased rates of clearance (Ref. Reference Mosley6). A more recent meta-analysis revealed the cumulative proportions achieving clearance were 19.8, 27.9, 36.1 and 37.1% at 3, 6, 12 and 24 months, respectively; thus individuals who had not cleared HCV spontaneously by 12 months were unlikely to do so. Again, increased rates of clearance were seen in females and with genotype 1 infection, as well as in those who were symptomatic. HIV co-infection, the absence of HBV co-infection, black or non-indigenous race, older age and problems with addiction to alcohol or drugs were all associated with reduced clearance rates (Ref. Reference Aisyah7). There is some evidence that HCV diversification during the acute phase of infection is associated with reduced spontaneous clearance rates (see below; quasispeciation). The observation that spontaneous clearance is rare once a patient has entered the chronic phase of infection, defined by convention as 12 months, is supported by a large population-based cohort study, in which 16 of 435 (3.7%) HCV RNA-positive individuals became RNA-negative during a mean follow-up period of 7.2 years (Ref. Reference Watanabe8) (an incidence of spontaneous clearance of 0.5%/year/person). No, or minimal, derangement of liver enzymes and an absence of liver ultrasound abnormalities were associated with spontaneous viral elimination.
A key region of genetic polymorphism associated with spontaneous viral clearance has been identified within the IFN-λ gene cluster. The first three members of this family of anti-viral proteins were originally termed IL29, IL28A and IL28B, but were subsequently renamed as IFN-λ1, IFN-λ2 and IFN-λ3 to better reflect the interferon-like nature of these proteins (Ref. Reference O'Brien, Prokunina-Olsson and Donnelly9). Initial genetic studies identified a non-coding single nucleotide polymorphism (SNP) rs12979860 that lies upstream of the IFN-λ3/IL28B gene that is associated with rates of spontaneous clearance of 50–60% of those homozygous for the major allele (C/C), compared with just 10–20% for individuals with the C/T or T/T genotypes (Refs Reference Thomas10, Reference Tillmann11). The mechanistic basis of this associated remains only partially resolved; indeed such was the impact of the initial discovery and the link to what was then termed IL28B, that the favourable genotypes at this locus are still frequently referred to using the nomenclature ‘IL28B’. Nevertheless, a key advance came with the recognition that a SNP in tight linkage disequilibrium with rs12979860, rs11322783 (originally designated as ss469415590), introduces a dinucleotide frameshift mutation that leads to the production of a functional additional IFN-λ family member, IFN-λ4, that is not expressed in individuals lacking the SNP and that plays a role in anti-viral defence (Refs Reference O'Brien, Prokunina-Olsson and Donnelly9, Reference Prokunina-Olsson12).
A similar correlation has also been reported between possession of the protective variants of rs12979860 and viral clearance in those undergoing treatment. This was first reported in those treated with IFN-α and ribavirin (Ref. Reference Ge13). SNPs near the IFN-λ3 gene associated with favourable treatment response are associated with higher levels of the hepatocyte-specific micro-RNA miR-122, which is important for stability and propagation of HCV RNA (Ref. Reference Su14). miR-122 is itself a target of IFN-induced genes, suggesting one possible mechanistic link between variants of this genotype and response to IFN treatment (Ref. Reference Hayes15). Similarly, high endogenous expression of IFN-induced genes has been associated with the same genotype and shown to predict a poor response to IFN treatment (Refs Reference Dill16, Reference Feld17).
In the era of DAAs, with greatly improved treatment response rates, the relevance of SNPs near the IFN-λ3 gene is less clear with any potential effect sizes likely to be much smaller given the higher baseline response rates. Early reports do indeed suggest that these polymorphisms retain a weak association with the response to combination treatment based on the protease inhibitor (PI) telaprevir with IFN-α and ribavirin, the clinical relevance of which might be to determine eligibility for shorter therapy durations (Ref. Reference Tsubota18), but this treatment combination has been superseded by more effective therapies where the effects of these polymorphisms remain to be determined.
Chronic infection
Chronic infection with HCV remains one of the leading causes of end-stage liver disease, hepatocellular carcinoma and liver-related death in the developing world. Reported rates of progression to cirrhosis vary, ranging from 2–3 to 51% over two decades (Refs Reference Kenny-Walsh19, Reference Wiese20, Reference Wiese21, Reference Tong22), with perhaps the most accurate estimates around 10–20% over 20–30 years (Ref. Reference Westbrook and Dusheiko4). Such wide ranges in progression rates are likely to reflect referral bias (Ref. Reference Fu23). Furthermore, the heterogeneous nature of infected populations also influences these very disparate estimates, with age, age at infection, gender, obesity, type 2 diabetes mellitus and alcohol consumption all having a bearing on the eventual outcome. Thein et al. performed a systematic review of 111 studies, including 33 121 patients, which demonstrated that whilst the overall progression to liver fibrosis in chronic HCV infection is influenced significantly by duration of infection, progression occurs in a non-linear manner between different stages of liver fibrosis (Ref. Reference Thein24).
The critical role of host immunity in determining controlling viral replication in chronic HCV is manifest in those immunosuppressed following HIV infection or following liver and other organ transplantation. Combined infection with HIV is associated with less spontaneous clearance, higher HCV RNA viral load and more rapid progression of liver disease. It also doubles the risk of mother to child transmission (Ref. 25).
Viral structure
The elucidation of the structure of the HCV represents a key starting point for the development of anti-viral therapies and was the subject of a recent review (Ref. Reference Adams, Pirakitikulr and Pyle26). Cryo-electron microscopy reconstructions and RNAse mapping were initially used to explore the HCV genome, followed more recently by high-resolution X-ray and nuclear magnetic resonance spectroscopy. Additionally, crystallographic studies of key viral proteins led to early predictions of possible functions and interactions, as well as identification of potential drug targets (Ref. Reference Dibrov27). Validation of these observations through mutational analysis in cell-culture models enabled assignment of functional roles of the structural elements.
Both in vitro and in vivo models have been used to study the HCV lifecycle, most notably using a replicon system. This is a self-replicating section of viral RNA derived from the viral genome and which contains viral non-structural genes that are critical for viral genome replication whilst structural proteins have been deleted. These systems have enabled assessment of the role of each part of the viral genome (Ref. Reference Lohmann28).
Lastly, in recent years, selective 2′-hydroxyl acylation analysed by primer extension (SHAPE) has been used, alongside RNA-sequencing to model the structures of different HCV genotypes (Refs Reference Mauger29, Reference Pirakitikulr30). Together, this array of laboratory studies has enabled identification of structural elements within conserved regions of the viral genome, for which the influence on viral replication and evasion of the host immune response could be explored. Importantly, this understanding has underpinned the development of therapeutic targeting of key viral structures, as discussed later in this review.
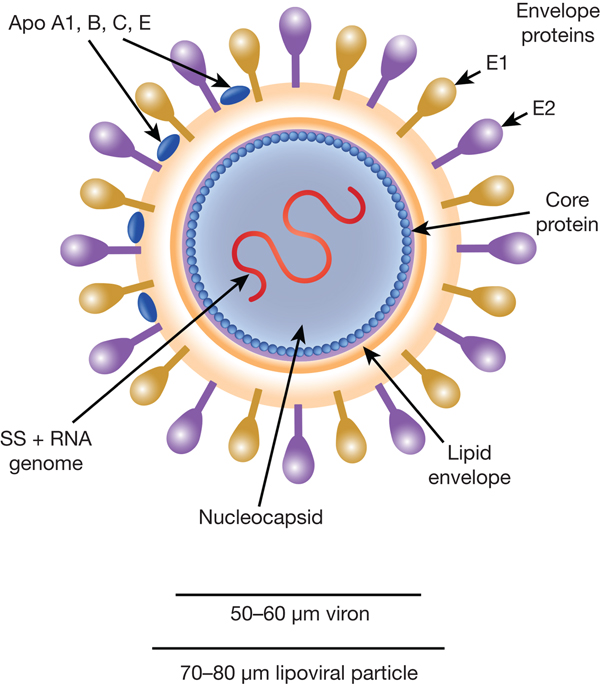
HCV particles are 40–70 nm in diameter (Ref. Reference Wakita31) and associate closely with host cell lipids, including lipoproteins (Fig. 1). This results in viral particles of low density and considerable heterogeneity. Electron micrographs of particles confirm this heterogeneity and show rather amorphous, spherical structures (Ref. Reference Merz32). At their core, these particles harbour the positive-sense, single-stranded 9.6 kb RNA genome of HCV. The genome contains a single open reading frame (ORF) encoding a viral core protein and envelope glycoproteins (E1 and E2) as well as a series of non-structural proteins. The ORF is flanked by 5′ and 3′ un-translated regions (UTRs), which contain cis-acting RNA elements.
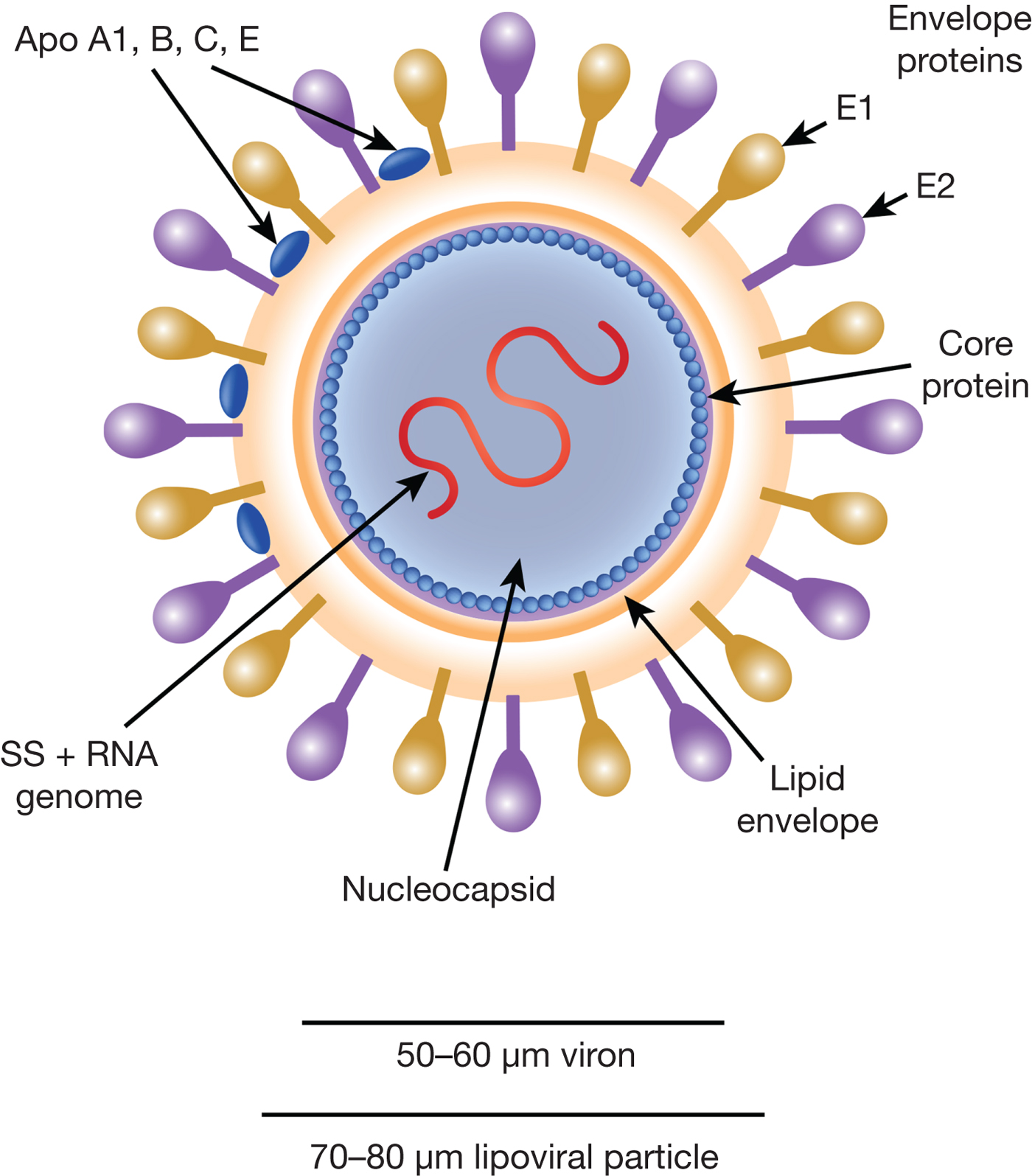
Fig. 1. The HCV viral particle. The HCV viron is a 50–60 µm structure encapsualted in a lipid-rich envelope to form a 70–80 µm lipoviral particle, the surface of which is coated in the envelope proteins E1 and E2 (see text).
Structure of the 5′ UTR
The 5′-UTR is of critical importance for the regulation of the viral life cycle. It has been studied extensively in terms of structure and function, in particular in terms of the critical interactions with the host cell ribosomal apparatus that initiate and perpetuate viral RNA translation. The 5′-UTR contains 340 nucleotides in four domains. Domain I is a short stem loop flanked by a binding site for the micro-RNA miR-122, whilst domains II–IV constitute an internal ribosome entry site (IRES) (Ref. Reference Adams, Pirakitikulr and Pyle26). Domains II and III form large stem-loops; domain IV forms a short unstable stem that includes the start codon. The domains are flexible, which enables the IRES to recruit the ribosomal 40S subunit and initiate translation of viral genes. To do this, the 40S subunit complexes with eukaryotic initiation factors eIF2 and eIF3 to form a higher order translational initiation complex, the 48S pre-initiation complex, which in turn binds with the 60S ribosomal subunit to form the functional 80S complex required for translation.
Domain III binds in an elongated manner at the back of the 40S and provides a stable platform for translation through direct interaction of its subdomains (IIIa–f) with mRNA in the mRNA channel. The interaction between the 40S subunit and domain III is critically dependent upon strong bonds formed between expansion segment 7 (ES7) of the 18s rRNA component of the 40S subunit on the one hand and subdomains IIId and IIIe on the side of the virus. Base pair mutations on domain III reduce IRES activity, but this is restored by compensatory mutations in ES7 (Ref. Reference Matsuda and Mauro33). eIF3 binding is mediated by subdomain IIId and the junction IIIabc (Refs Reference Perard34, Reference Quade35). Structural studies have observed that this interaction displaces eIF3 from its canonical binding site on the 40S subunit, in so doing disrupting the ability of eIF3 to initiate translation of host mRNA and so subverting host cell machinery towards viral mRNA translation (Ref. Reference Hashem36).
Domain II of the 5′-UTR is an independent L-shaped structure, both in free solution and when bound, which reaches across the 40S subunit, into the inter-subunit space. It induces a conformational change in the 40S, with a possible role in the proper positioning of the mRNA on the ribosomal complex during translation (Ref. Reference Spahn37). Whilst domain III alone is sufficient for 48S assembly, domain II has been shown to control 80S ribosome formation downstream of 48S assembly. In particular, domain II appears to be required to promote GTP hydrolysis by eIF5 and subsequent eIF2/GDP release from the 48S complexes, two key steps in the formation of the full 80S ribosome formation (Ref. Reference Locker, Easton and Lukavsky38). Finally, domain IV must unfold in order for the start codon to be positioned in the appropriate site on the 40S subunit for translation to begin.
Regulation of HCV translation occurs within the 5′-UTR through the action of host micro-RNA. In particular, in contrast to the typical down-regulation of protein expression from target mRNAs through limited base-pair interactions, the hepatocyte-specific miR-122 has two partially overlapping binding sites at the terminus of the HCV 5′-UTR, with extensive base pairing outside the seed sequence (Ref. Reference Mortimer and Doudna39). The binding of miR-122 to the virus results in a stable ternary complex and so enables HCV RNA to sequester miR-122 from endogenous cellular mRNA targets (Ref. Reference Luna40). A 3′ overhang (6 miRNA nucleotides) is created when the miR-122 binding site 1 extends across domain I, which may protect the HCV genome from cellular exonucleases (Ref. Reference Wilson and Sagan41); the effect of the miR-122 HCV interaction on viral infectivity remains unknown.
IRES trans-acting factors
Several host proteins are recruited to the IRES to mediate activity, known collectively as IRES trans-acting factors (ITAFs). The exact mechanisms by which ITAFs modulate IRES activity remain unknown. ITAFs were the subject of a recent excellent review by Niepmann (Ref. Reference Niepmann42), so are not covered in detail here, with the exception of Human La protein, which binds to the HCV RNA through a β-turn and stimulates IRES-dependent translation. Of potential therapeutic interest, the construction of a synthetic 24-mer peptide (LaR2C), derived from the C terminus of the RNA recognition motif of the La protein, can compete with cellular La protein at the binding site and interfere with the formation of a functional translation initiation complex (Ref. Reference Pudi, Ramamurthy and Das43).
A cyclic 7-mer peptide, derived from LaR2C, has also been shown to mimic the La protein β-turn and inhibit IRES-mediated translation of HCV, as well as inhibiting the growth of HCV replication in a cell culture model. This 7-mer sequence is conserved only in humans and chimpanzees providing a possible explanation for the lack of infectivity of HCV in small animal models, which in turn limits the available platforms for viral study and therapeutic development. The generation of a chimeric La protein harbouring a human La β-turn enhanced HCV replication in murine cells and may suggest a possibility of creating a mouse model of HCV infection (Ref. Reference Kumar44).
The open reading frame
The ORF produces a ~3000 aa polypeptide which is co- and post-translationally cleaved into viral proteins (Fig. 2). The core protein is a highly basic 191 amino acid RNA-binding protein that forms the viral capsid (Ref. Reference Yasui45). The E1 and E2 envelope proteins are heavily glycosylated and play essential roles for cell entry through interaction with cell surface receptors including CD81 (Ref. Reference Pileri46) and SCARB1 (Ref. Reference Scarselli47). They are highly immunogenic, but extreme genetic variation has limited the development of successful vaccines (Ref. Reference Tarr48).

Fig. 2. HCV viral proteins. After translation, HCV viral proteins sit with varying relationships to the ER membrane. During maturation, the core protein is cleaved by signal peptide peptidase. The envelope proteins E1 and E2 are cleaved from each other and from the p7 ion channel by ER signal peptidase to lie embedded within the ER membrane where they are further heavily glycosylated. NS2 also lies predominantly within the membrane, formed of dimeric subunits and is cleaved from NS3 by the autoproteolytic action of the NS2/NS3 complex. The N-terminus of NS3 is a serine protease and associates with the transmembrane NS4A serine protease which acts as a co-factor. The NS3/4A protease cleaves the remainder of the non-structural proteins from each other. The C-terminus helicase domain of NS3 sits in the cytosol whilst NS4B is embedded within the ER membrane. NS5A is anchored in the ER membrane but the bulk of the protein sits within the cytosol where it plays critical roles in MMV assembly and viral replication. NS5B is the viral RNA-dependent RNA polymerase (RdRp) and is anchored in the ER membrane by a C-terminal transmembrane segment (Ref. Reference Moradpour, Penin and Rice60).
Reading from the N-terminus of the polypeptide chain (corresponding to the 5′ end of the ORF), the ORF then encodes the p7 ion channel (Ref. Reference Griffin49), followed by a series of non-structural proteins (Fig. 2). The NS2/3 protein comprises NS2 and the N-terminal of NS3, forming a highly hydrophobic protease that functions through autoproteolysis to cleave the viral polypeptide between NS2 and NS3 (Ref. Reference Welbourn and Pause50).
NS3 itself has both serine protease (N-terminus) and helicase (C-terminus) activity, whilst NS4A functions as a co-factor for NS3 protease activity (Ref. Reference Shiryaev51). These activities are critical for viral replication rendering NS3/4A a key DAA target. Of the two mechanisms, inhibition of protease activity has so far shown more clinical success. The protease appears relatively non-specific and known host cellular targets include regulators of the IFN response (Refs Reference Meylan52, Reference Li53).
NS4B is a less well-characterised protein that sits within the endoplasmic reticulum (ER) and is thought to facilitate the assembly of membrane structures, termed multi-membrane vesicles (MMV), that facilitate viral RNA replication (Ref. Reference Moradpour54).
NS5A is a ~450 amino acid protein that lacks apparent enzymatic activity, but sits anchored within the ER membrane by an amphipathic α-helix. Further major domains include domains D1, D2 and D3, linked by short, easily cleaved linker sequences (Ref. Reference Tellinghuisen55). The whole protein undergoes extensive phosphorylation which in turn impacts viral replication. Functions of NS5A have been proposed from selective deletion studies of individual domains, which appear to affect in turn viral replication (D1 and to a lesser extent D2), binding to host cell proteins (D2 and D3) with resultant subversion of host cell signalling and the interferon response (Refs Reference Yang56, Reference Reiss57) and binding to viral RNA to protect from degradation and aid viral assembly (D1 and D2) (Refs Reference Bartenschlager, Lohmann and Penin58, Reference Schmitz and Tan59). The extensive range of NS5A functions makes it attractive for therapeutic targeting, even in the absence of enzymatic function.
Finally, NS5B is the viral RNA-dependent RNA polymerase (RdRp) which is critical for viral RNA synthesis (Ref. Reference Moradpour, Penin and Rice60). At the core of the enzyme lies a highly conserved catalytic domain. In the inactive state, this lies buried against the membrane and NS5B must undergo a conformational change to open this site to generate dsRNA (Ref. Reference Mosley61). Thus agents targeting NS5B may do so either by blocking the active site or by preventing structural changes within the protein (discussed further below).
In addition to these coding roles, the ORF has highly specific RNA structures (Refs Reference Mauger29, Reference Pirakitikulr30), some of which are conserved across all genotypes, but divergent regions account for differences in pathogenicity between genotypes. In particular, stem loops formed by the 5′ end of the ORF within the region encoding the core protein are required for replication of the intact genome (Ref. Reference Vassilaki, Boleti and Mavromara62). Similarly, at the other end of the ORF, within the region encoding NS5B, there is a set of stem-loops that interact with RNA motifs in the 3′ UTR, and that are also essential for HCV replication.
The 3′-UTR
Three regions of the positive strand 3′-UTR have been identified. A variable region of approximately ~40 nucleotides near the stop codon forms two stem-loops which overlap with the 3′ terminus of NS5B. Next, a flexible polyU/UC tract of between 30 and 80 nucleotides is thought to provide a platform for recruitment of host or viral proteins. Finally, three highly conserved stem loops at the terminus known as the 3′ X-tail are essential for viral replication (Ref. Reference You and Rice63), with one of the tail stem loops (SL2) forming a tertiary structure with an RNA element in the NS5B coding region (5BSL 3.2) (Ref. Reference Sagan, Chahal and Sarnow64).
The structure of the 3′-UTR of the negative strand is poorly characterised. However, alongside sections of the positive strand, it is likely to direct the HCV RNA polymerase for replication. NS5B polymerase has been noted to initiate replication more efficiently from the negative-strand 3′-UTR than the positive, leading to the suggestion that this creates a more robust complex (Ref. Reference Reigadas65) and may explain the relative abundance of positive-strand over negative-strand genomes within infected cells (Ref. Reference Quinkert, Bartenschlager and Lohmann66).
Viral life cycle
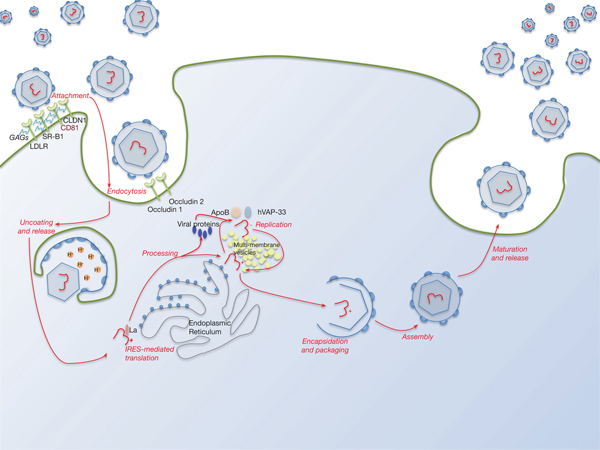
Lipid pathways are essential to the viral life cycle and by modulating their production in the host cell, HCV is able to make lipoproteins more effective for viral production, propagation and persistence. The virus is able to circulate through the blood stream as a highly lipated lipoviral particle (LVP) and then utilises the lipoprotein pathway for cell entry and virus assembly (Fig. 3). This may also be involved in RNA replication. The resemblance of LVP with VLDL/LDL may also contribute to viral immune evasion strategies (Refs Reference Grassi67, Reference Vercauteren68). Several lipid abnormalities have been associated with chronic HCV infection, including liver steatosis in those with genotype 3, hypobetalipoproteinaemia and hypocholesterolaemia (the latter reverses with successful antiviral therapy) (Ref. Reference Grassi67).

Fig. 3. The HCV lifecycle. Infection of host cells begins with attachment of the virion to the host cell surface in a process involving host cell surface coreceptors including CD81, and facilitated by claudin 1 (CLDN1), the SR-B1 scavenger receptor and low-density lipoprotein receptor (LDLR). Glycosaminoglycans (GAGs) decorating these proteins play a critical role in this interaction. After viral attachment, the process of endocytosis proceeds in a clathrin-dependent manner with key roles played by host occludins 1 and 2. After vesicle acidification, release of the virus into the cytoplasm occurs. Translation and processing of the viral proteins takes place on the endoplasmic reticulum, with assembly of the translational apparatus around an internal ribosome entry site (IRES) formed within domains II–IV of the 5′-UTR (see text). Viral replication is driven by the NS5B RNA polymerase and takes place on a lipid-rich multi-membrane vesicles (MMV), the assembly of which requires is facilitated by NS4B, whilst hVAP-33 plays an important role in assisting assembly of the replication complex on the MMVs. Finally, viral encapsulation, packaging, assembly and release take place in a poorly characterised sequence that involves NS5A amongst other proteins.
HCV can also circulate as free virions bound to immunoglobulins (Ref. Reference Andre69). The interaction of infectious viral particles with both lipoproteins and immunoglobulins partially explains why they have a lower buoyant density in comparison to other enveloped positive-strand viruses (Ref. Reference Lindenbach70). This property is also thought to have an effect on increased infectivity (Ref. Reference Yin71).
Hepatocytes are the main target for infection and unusually for viral infection, many receptors have been proposed to enable viral entry, but these are not covered in detail in this review. In HCV-infected cells, non-structural viral proteins including NS5A localise to the ER as well as to lipid droplets and to MMV that host RNA replication complexes. The MMV in particular are enriched for NS3-NS5B and viral RNA. Recent studies have used crystal structures to model the events during replication. These suggest that NS5B non-canonical polymerase elements (a C-terminal membrane which acts as an anchor and a β-loop insertion point) are buried within the encircled active site of the enzyme. During de novo initiation of translation, the 3′ end of the RNA template and incoming plus priming nucleotides enter the active site, resulting in a dinucleotide primer. This precedes the primer initiation assembly. As the primer extends, the tension displaces the β-loop and C-terminal residues allowing the RNA duplex to exit; they are finally expelled enabling the polymerase to transition into a highly processive elongation mode (Ref. Reference Appleby72).
The transportation of these genomes to sites of viral assembly is poorly characterised. However, the viral genome and structural proteins are packaged in the MMV prior to maturation and eventual release; it is thought that NS5A has a role in co-ordinating packaging (Ref. Reference Yin71). The cellular vesicle membrane transport protein hVAP-33 (the human homologue of the 33-kDa vesicle-associated membrane protein-associated protein) binds to two different domains on NS5A and NS5B. A number of proteins involved in membrane fusion interact with hVAP-33, including those associated with the lipid raft, and thus hVAP-33 is thought to be crucial to the replication complex on cholesterol-rich lipid rafts (Ref. Reference Gao73). Indeed, down-regulation of h-VAP proteins has been shown to inhibit HCV replication, thus producing an antiviral effect, which is more pronounced when multiple genes are down-regulated simultaneously such as h-VAP-33 in combination with La antigen (Ref. Reference Mandal, Ganta and Chaubey74).
After release, the estimated half-life of a HCV virion is around 2.7 hs, with production and clearance of an estimated 1012 virions per day during active infection (Ref. Reference Neumann75).
Genetic diversity
HCV isolates cluster into seven different genotypes with a further 67 subtypes. Sequence variability between the main genotypes can exceed 30%, whilst the subtypes show variability exceeding 15%, based upon analysis of a variability within a small region of NS5B (Ref. Reference Smith76). In the West the majority of infections are attributable to subtypes 1a, 1b and 3a.
The high genetic variability of the virus is related to high replication rates in addition to a lack of proof reading of the viral NS5B RdRp, leading to an estimated 10−4 substitutions per replication cycle (Ref. Reference Bartenschlager and Lohmann77). Within an individual, the genomic population itself is heterogeneous, with a collection of microvariants derived from the predominant sequence referred to as ‘quasispecies’. This variation may account for the significant proportion of viral genomes that are defective (Ref. Reference Martell78).
Immunity and defence against HCV infection
The immune response to HCV is a complex story of interplay between host defences and viral strategies of evasion. This has been recently well reviewed elsewhere (Refs Reference Dustin79, Reference Park and Rehermann80) and is outlined in brief here.
The physically compact and highly structured nature of the HCV viron is the first-line defence against degradation by the immune system. HCV does not cause a direct cytopathic effect on host cells, but instead the virus is thought to cause alterations in the immune system and metabolic pathways.
The human host has numerous mechanisms to defend against viral infection. ‘Restriction factors’ are a collection of proteins/peptides that have direct antiviral activity, affecting almost all stages of the HCV life cycle. Subsequently, coordinated actions of components of innate and adaptive immune systems include the activation of natural killer cells, processing of viral antigens by dendritic cells and establishment of B- and T-cell responses against those antigens.
HCV behaves differently in mild as opposed to severe disease; in particular, there is conflicting evidence as to whether high-diversity quasispecies are associated with lower or higher disease severity, or indeed if there is any relationship at all. What has been recognised is that in immunosuppressed patients with progressive liver disease, quasispecies diversity decreases over time (Ref. Reference Qin81). Supporting this consistent finding, one study observed that the progression of HCV was associated with homogenisation of HCV quasispecies in treatment-naïve, immune-competent patients over time, with HCV hypervariable region diversity showing significantly lower rates of change in those with severe disease (Ref. Reference Sullivan82).
Treatment
The switch from acute to chronic infection is not well delineated, with duration of acute viraemia varying considerably between individuals. As many patients are unaware of their acute infection, intervention at this point is not common; but treatment based on recombinant type-I interferon has long been used in the acute phase to reduce the risk of chronic hepatitis, with high rates of viral clearance observed in those treated for 24 weeks or longer. Whilst response rates are lower in those treated later than 20 weeks, treatment is not usually commenced until at least 12 weeks post-infection to enable assessment for natural clearance (Ref. Reference Westbrook and Dusheiko4).
Early treatment
In the mid-1990s, treatment for HCV became available in the form of subcutaneous interferon-α as part of a year-long regime which was poorly tolerated. Treatment success was defined as ‘sustained virological response’ (SVR), with the absence of detectable viraemia 24 weeks after completion of treatment. SVR rates were as low as 10–15% with such IFN-based regimens. Studies of these treatment regimens showed that infected cell death rate in the first 2 weeks of therapy correlated with status at 3 months and that there was an inverse correlation of infected cell death rate with baseline viral loads. Thus if viraemia persisted after 3 months therapy, an informed decision about the likely success of continuing treatment could be made by considering these factors (Ref. Reference Neumann75).
The subsequent introduction of a pegylated form of interferon-α with more favourable pharmacokinetics allowed weekly rather than thrice-weekly regimes. Alongside this, combination therapy with ribavirin, a guanosine analogue nucleoside inhibitor (with little intrinsic antiviral activity), led to improved efficacy with SVR rates of 40–50% but at the expense of worsening side effects.
In 2011, the first drugs targeting a specific viral component were introduced, in the form of the NS3/4A PIs telaprevir and boceprevir. In patients infected with HCV genotype 1, when combined with pegylated interferon-α and ribavirin, this treatment led to SVR rates around 75%.
Direct acting antivirals
Despite our limited, albeit expanding, knowledge of how the HCV replicates in infected liver cells, treatment has been revolutionised over the last few years through the discovery of DAAs. Targets for these include the NS3/4A protease, NS5A and NS5B RdRp (Table 1). DAAs are now widely used in combination therapies to treat all genotypes with SVR rates 12 weeks post-therapy of ~95%, almost irrespective of patient demographic or liver disease severity, although patients with decompensated cirrhosis seem to have lower rates of around 85% (Ref. Reference Vermehren83). Importantly, the side effect profile is much more favourable when compared to interferon- and ribavirin-based regimens, treatment duration is now considerably shorter, and the improved SVR rates are seen even in those who failed earlier treatments.
Table 1. Overview of current DAAs in HCV therapeutics

In a rapidly changing landscape of therapeutic options and evidence, DAA regime choices, the addition of ribavirin and duration of treatment are all dependent on multiple factors including prior treatment, disease severity and genotyping. Guidelines vary between countries and will not be covered in full here.
HCV protease inhibitors
The HCV protease is a heterodimer within infected hepatocytes, formed from the combination of viral NS3 and NS4A proteins (Ref. Reference Poveda84). HCV PIs interact with the protease substrate binding site, thereby preventing HCV cleavage and subsequent generation of the non-structural proteins. The catalytic site of this protease exists in a shallow groove, which requires small molecule inhibitors to rely on a small number of specific interactions to achieve tight binding with the enzyme. As a result, there was a low genetic barrier to resistance and cross-resistance in the first generation of PIs, although this was greatly improved in the second (Refs Reference Jacobson85–Reference Manns87). Those in current use include simeprevir, paritaprevir and grazoprevir. Recently licensed agents that are also increasingly used in this class include glecaprevir and voxilaprevir, with several new agents in this class in late phases of clinical development and licensing.
NS5A inhibitors
The exact mechanism of action of the NS5A inhibitors remains unknown as a result of our lack of understanding of how NS5A regulates viral replication, assembly and particle release. However, these inhibitors have broad genotypic coverage, and despite their low barriers to resistance, they are included in nearly all current treatment regimes (Refs Reference Pawlotsky1, Reference Zeuzem88). Currently approved NS5A inhibitors include daclatasvir, ledipasvir, ombitasvir, elbasvir, pibrentasvir and velpatasvir. Again, several novel agents in this class are in late phase development.
NS5B polymerase inhibitors
As discussed above, NS5B is the RdRp of the HCV replication complex. Inhibitors of this polymerase may fall into different classes depending on their mode of action. Nucleoside analogue inhibitors mimic the substrates of the polymerase and are thereby incorporated into the nascent RNA chain but ultimately result in chain termination (Ref. Reference Gentile89). Non-nucleoside analogue inhibitors bind at a range of sites including those within and outside the active site of the polymerase; these allosteric binding sites include those within the Palm and Thumb domains (Ref. Reference Gerber, Welzel and Zeuzem90). Binding of non-nucleoside analogue inhibitors to NS5B likely inhibit conformational changes necessary for effective function of the protein.
There are currently two approved agents in this class. Sofosbuvir is a nucleoside analogue inhibitor. Dasabuvir is a non-nucleoside analogue inhibitor and binds at palm 1 effectively blocking viral RNA polymerase function. Sofosbuvir is pangenotypic, with a high barrier to resistance, whilst dasabuvir is used for the treatment of genotypes 1a and b, with naturally occurring mutations and low-level resistance.
Treatment resistance
As already alluded to, efficacies of different DAAs are affected by baseline, naturally occurring mutations within the HCV genome (Table 1). Naturally occurring mutations confer reduced susceptibility to either specific drugs, or entire DAA classes (Ref. Reference Sarrazin and Zeuzem91) and are generally attributable to changes in the conformation of the binding site of the DAA to the HCV protein. HCV polymorphisms are referred to according to standard nomenclature referencing the amino acid change in the viral protein. For example, S282T implies that the serine amino acid found in the wild-type position 282 has been replaced by a threonine residue, although the second letter after the amino acid position may be left blank to imply an unspecified change to any amino acid other than the original parent sequence (Ref. Reference Weisberg and Jacobson92). Notable polymorphisms include Q80K, found in 19–48% of HCV genotype 1a, which is associated with reduced susceptibility to simeprevir, and D168Q, found in nearly all genotype 3 genomes; conferring to resistance to most PIs (Refs Reference Poveda84, Reference Paolucci93). Both of these substitutions introduce structural changes in NS3.
In particular, some substitutions are associated with improved viral replication in the presence of DAAs (Table 1). These so-called viral resistance-associated substitutions (RAS) may be present even prior to DAA initiation or may occur spontaneously during viral replication but are usually found in only minor populations of the quasispecies, as a result of conferring less fitness than the wild-type virus. Following successful inhibition of the wild-type virus by DAAs, this relative fitness can be perturbed, resulting in positive selection of the resistant quasispecies. In reality, the impact of these polymorphisms may be negligible in combination regimes where other compounds retain full activity.
The S282T substitution described above confers resistance to sofosbuvir in vitro through alterations in NS5B structure. It is not observed in untreated patients, but may be detected in 1% of patients following sofosbuvir-based regimes. A recent study revealed that the substitution was unfit in vivo and would disappear in most patients in the absence of drug selection pressure. As such retreatment with sofosbuvir should be possible (Ref. Reference Gane94). Thus, the outgrowth of viral populations containing RAS is dependent on genetic barriers to resistance, levels and duration of drug exposure, viral fitness and the prevalence of naturally occurring mutations (Ref. Reference Lontok95).
Both drug- and class-specific RAS are observed and classified further according to their ability to restrict the action of the antiviral agent. Thus, a particular RAS is considered clinically significant if it confers at least a two- to fivefold increase in the concentration of drug required to inhibit replication by 50% (EC50) compared with the wild-type virus using the in vitro replicon system (Ref. Reference Pawlotsky96). The baseline RAS attenuate the efficacy of DAAs but treatment failures are inducing the emergence of highly resistant RAS with EC50 changes of up to 100-fold that of the wild-type replicon, emphasising the wisdom of combination therapies.
High frequencies of RAS to NS5A inhibitors have recently been seen in genotype 3. In particular, paired substitutions at A30K and L31M in all patients with genotype 3 subtypes b and g have been reported. In vitro analysis suggests this is associated with inherent resistance to all approved NS5A inhibitors for genotypes 3b and 3g (Ref. Reference Smith97). Similar naturally existing RAS to NS5A inhibitors have been described for other rare HCV genotypes (Refs Reference Welzel98, Reference Ordeig99).
Sequencing
Experience in HIV medicine has shown the devastating effects that anti-viral resistance can have on response to treatment. Therefore, where available, baseline HIV viral genome sequencing for resistance is now routine practice. A similar model of practice has not been adopted in the management of HCV. In part, this is due to the excellent rates of viral cure now being achieved and the ability to switch regime or increase treatment duration to overcome resistance, as well as to the increased costs associated with resistance testing.
There is also yet to be a consensus on sequencing protocols to detect RAS. A recent systemic review highlighted the diversity of sequencing practice, notably that current protocols do not cover all clinically relevant RAS and remain genotype-1 centric (Ref. Reference Bartlett100). Population-based sequencing only identifies viral populations if they represent approximately 15% of quasispecies, whilst deep sequencing can identify 0.5–1%. However, these minor variants do not yet appear to have clinical significance and thus more superficial population-based sequencing appears sufficient to identify the more relevant resistant variants that predict treatment failure.
Retreatment, reinfection and the role of treatment programmes
In the proportion of patients (5–15% dependent on regimen and treated population) who fail to eradicate HCV infection, RAS are generally the cause. The low barrier to resistance of the NS5A inhibitors is the principle challenge due to persistent RAS, in contrast to RAS in NS3/4A inhibitors that are progressively replaced by wild type within months. In this context, combination treatments are recommended in current guidelines, including the use of the NS3/4A inhibitor glecaprevir, with its high barrier to resistance, with the NS5A inhibitor pibrentasvir. A similar rationale underlies the alternative combination of the NS3/4A inhibitor voxilaprevir with the NS5A inhibitor velpatasvir and the NS5B inhibitor sofosbuvir. Importantly, this combinatorial approach has been shown to succeed even in the context of previous DAA-induced RAS-related treatment failure with SVR rates in excess of 90% (Ref. Reference Bourliere101). An alternative approach to overcoming the threat to DAA efficacy posed by RAS is to prolong treatment duration in combination therapy with ribavirin therapy (Ref. Reference Feld102).
Re-infection has long since been recognised, particularly in PWID and men who have sex with men. Prior to the introduction of DAAs, this was typically restricted to those who had cleared acute hepatitis C. Several health models have shown that even modest increases in successful treatment of HCV infection among PWID can decrease prevalence and incidence (Ref. Reference Panel103). However, there is also an expectation that there will be, if only temporarily, a rise in re-infection rates following successful treatment, particularly in the high-risk groups (Ref. Reference Martin104). Epidemiologically, sequencing will be critical to establishing rates of re-infection. For example, systematic sequencing of post-treatment samples has confirmed the emergence of pre-existing minor variants rather than re-infection (Ref. Reference Abdelrahman105).
The future
The success of current anti-viral therapies to treat HCV may be short lived if resistance and genotypic diversity continue to develop indicating a need for continued development of novel agents; in this regard, RNA structures are natural targets for such approaches. Given that the 5′-UTR has been best characterised, progress has already been made in targeting this region, including disruption of miR-122 binding (Ref. Reference Adams, Pirakitikulr and Pyle26). Viral variability has made vaccine development challenging, just as it has for the development of antiviral agents. Early phase I study vaccine studies using recombinant HCV E1/E2 EnvGPs were disappointing (Ref. Reference Kwon106). However, recent work using T-cell simian adenoviral vectored vaccines, which encode genetic segments conserved between all major genotypes, has been promising (Ref. Reference von Delft107).
An additional set of challenges come with the need to ensure wide application of curative strategies for all infected patients, not just those in the clinics, and in so doing to ensure these regimens are available worldwide. In November 2018, a royalty-free licence agreement was signed between the Medicines Patient Pool and AbbVie for glecaprevir/pibrentasvir (G/P), which will enable the development and manufacturing of generic medicines containing G/P in 99 low- and middle-income countries (Ref. 108). The availability of the most effective WHO recommended pan-genotypic regime at an affordable price should hopefully enable treatment scale up to work towards elimination targets set by the WHO (Ref. 109). Even in countries with universal reimbursement for DAAs, challenges remain. Australia, for example, successfully treated 17% of their population with HCV in 1 year, but is struggling to reach vulnerable groups such as those who inject drugs (Ref. Reference Richmond and Wallace110). Similar barriers have hampered efforts to reach vulnerable groups in England (Ref. 111).
The story of the move from HCV discovery to effective HCV treatment is indeed a remarkable one. The development of a cure for HCV simply was not foreseen, even a decade ago. Although work to understand all aspects of virus biology and develop strategies for overcoming RAS remain, the key questions now are whether and for how long HCV will remain a significant global healthcare burden – and this must rest in the domain of policy and public health.
Author ORCIDs
Lucy Rivett, 0000-0002-2781-9345; Graeme Alexander, 0000-0002-9713-1394
Financial support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Conflict of interest
None.