
Abstract
A shift of the delicate balance between apoptosis and survival-inducing signals determines the fate of neurons during the development of the central nervous system and its homeostasis throughout adulthood. Both pathways, promoting or protecting from apoptosis, trigger a transcriptional program. We conducted whole-genome expression profiling to decipher the transcriptional regulatory elements controlling the apoptotic/survival switch in cerebellar granule neurons following the induction of apoptosis by serum and potassium deprivation or their rescue by either insulin-like growth factor-1 (Igf1) or pituitary adenylyl cyclase-activating polypeptide (Pacap). Although depending on different upstream signaling pathways, the survival effects of Igf1 and Pacap converged into common transcriptional cascades, thus suggesting the existence of a general transcriptional program underlying neuronal survival.
Similar content being viewed by others

Introduction
Neuronal apoptosis represents an intrinsic suicide program by which a neuron orchestrates its own destruction. It is characterized by specific morphological and biochemical events, including fragmentation of nuclear DNA, breakdown of the cellular cytoskeleton and the bulging out of the plasma membrane (blebbing), which may lead to the detachment of the so-called apoptotic bodies.1 During normal nervous system development, physiologically appropriate neuronal loss contributes to a sculpting process that removes approximately one-half of all neurons born during neurogenesis.2 Neuronal loss subsequent to this developmental window is physiologically inappropriate for most systems and can contribute to neurological deficits, for example, neurodegenerative diseases such as Alzheimer's and Parkinson disease.3 Hence, elucidating the molecular mechanisms underlying neuronal apoptosis may contribute to understanding the basis of developmental biology and human neuropathology.
Cerebellar granule neurons (CGNs) are the most abundant neuronal cell type in the mammalian brain and are used as a model, either in vivo or in vitro, to study neuronal apoptosis.4,5 Loss of neurotrophic supply and/or activity-dependent survival signals can induce apoptosis in CGNs. The relative contribution of these components correlates to the neuronal type and age. During early stages of postnatal development, it is assumed that apoptosis of granule cells reflects the failure of these neurons to obtain adequate amounts of specific neurotrophic factors.4 In the adult, mossy-fiber axotomy is followed by apoptosis of CGNs, highlighting the crucial role of afferent inputs on the survival of these cells.
Primary cultures of CGNs have been extensively utilized to examine the signal transduction mechanisms underlying neuronal apoptosis. In this in vitro paradigm, CGNs undergo rapid apoptotic cell death within 24 h after the removal of serum and lowering of extracellular potassium from 25 to 5 mM.5 The apoptotic process requires protein transcription and synthesis, becoming irreversible after the first 6 h following its induction.6 Before this ‘commitment point’, CGNs can be rescued by the activation of specific signal transduction pathways or by the treatment with specific neurotrophic factors. In our previous studies, we identified two important growth factors capable of preventing apoptosis of CGNs: insulin-like growth factor-1 (Igf1)5 and pituitary adenylyl cyclase-activating polypeptide (Pacap).7 The survival effects of these growth factors are mediated by different receptors and intracellular second messengers.5,7 Although these signaling pathways converge into the nucleus and regulate gene expression, the transcriptional program underlying neuronal survival is still unknown. In the present work, we carried out whole-genome expression profiling to investigate the rescue effects of Igf1 and Pacap in CGNs and identified crucial genes and pathways at the intersection of neuronal apoptosis and survival.
Results
Induction of apoptosis and rescue by Igf1 and Pacap
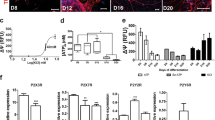
CGNs undergo apoptotic cell death after the removal of serum and lowering of extracellular potassium from 25 to 5 mM,5 and can be rescued by Igf15 and Pacap treatments.7 To confirm this in our in vitro paradigm and select the doses of Igf1 and Pacap having similar efficacy, we used three diverse methods to assess apoptosis and survival. Neuronal viability was assessed by counting the number of intact nuclei, whereas determination of oligonucleosomes generated by cleavage of nuclear DNA was performed with enzyme-linked immunosorbent assay (ELISA) and electrophoresis on a microchip device (Figures 1a–c). Forty-eight hours after the induction of apoptosis, neuronal viability of CGNs was ~32% of control and DNA fragmentation increased 3.5-folds (Figures 1a and b).

Induction of apoptosis in CGNs and rescue by Igf1 and Pacap treatment. (a) Cultured CGNs at 6 days ‘in vitro’ were switched into serum-free medium containing lower concentrations of extracellular K+ (5 instead of 25 mm) for an acute induction of apoptotic death. Forty-eight hours later, neuronal viability was assessed by counting the number of intact nuclei. Values for neuronal viability represents the mean±S.E.M. of four to eight determinations in two different experiments. Determination of oligonucleosomes generated by cleavage of nuclear DNA was performed with ELISA (b) and electrophoresis on a microchip device (c). Values for DNA fragmentation assay represent the mean±S.E.M. of four to eight determinations in two different experiments. P<0.001 versus K25 (*) or K5 (**) were determined by one-way ANOVA followed by Tukey post hoc test (a and b). (d and e) Genes differentially expressed in apoptotic CGNs (K5 versus K25) were defined as ‘Apoptotic related genes’. Genes differentially expressed in rescued CGNs (K5+Igf1 versus K5; K5+Pacap versus K5) were defined as ‘Survival related genes’. (f) Hierarchical cluster of genes related to apoptosis and rescue of CGNs. ARGs and SRGs were arranged in a dendrogram in which the pattern and length of the branches reflect the relatedness of the expression levels under four different experimental conditions. Green, black and red cells, respectively, are transcript levels below, equal or above the median abundance across all conditions. Color intensity reflects the magnitude of the deviation from the median. The Pearson correlation coefficient of SRG fold ratios (K5+Igf1/K5, K5+Pacap/K5) is 0.97.
Treatment with Igf1 or Pacap prevented most of CGNs from undergoing apoptosis and the maximal efficacy reached by 3.26 pm Igf1 was similar to that obtained with 100 nm Pacap (Figures 1a–c).
Whole-genome expression changes underlying apoptosis and survival
By using oligonucleotide microarrays, we monitored whole-genome expression profiles of CGNs after induction of apoptosis and following rescue by a maximal effective dose of Igf1 (3.26 pm) or Pacap (100 nm). To exploit the comprehensiveness of our data, we investigated changes on the level of individual genes and in functional gene groups.
Identification of differentially expressed genes
When gene expression profiles in control CGNs (K25) were compared with those of CGNs 6 h after induction of apoptosis (K5), 1212 genes, operationally defined as ‘Apoptotic Related Genes’ (ARGs), showed significant changes of gene expression (Figures 1d and e). By comparing gene expression profiles in CGNs 6 h after induction of apoptosis (K5) with those of apoptosis-rescued CGNs by treatment with Igf1 and Pacap, 1535 genes were found to be differentially expressed and were operationally defined as ‘Survival Related Genes’ (SRGs; Figures 1d and e). Among these, 968 and 1315 genes were differentially expressed after treatment with Igf1 and Pacap, respectively (Figure 1e).
Although the survival effects of Igf1 and Pacap are initiated by different receptors and activate a variety of intracellular second messengers,5,7 they were propagated by common transcriptional cascades and showed striking similarities. Indeed, 748 out of 1535 SRGs overcoming a stringent cutoff level were common to Igf1 and Pacap (Figure 1e). The Pearson correlation coefficient between Igf1 and Pacap for all 1535 SRGs was 0.97, further supporting the similarity of their genomic effects (Figure 1f).
A comprehensive picture of transcriptional changes associated with apoptosis and survival is shown in Figure 1f, where ARGs and SRGs (a total of 2293 genes) are grouped on the basis of similarity in their expression patterns with a hierarchical clustering method. The large impact of apoptosis and survival on the CGNs transcriptome, together with the remarkable concordance of Igf1- and Pacap-induced effects can be distinguished by the color matrix. The hierarchical cluster shows in an unequivocal manner that gene expression patterns of Igf1 and Pacap were highly similar to each other (Figure 1f).
Although our data represent the average gene expression from four replicates, we confirmed the reliability of the microarray data by quantitatively validating the differential expression of seven genes in each of the four experimental conditions using quantitative RT-PCR (Supplementary Table S2). Using RT-PCR, the pattern of gene expression from sample to sample closely paralleled that observed using microarray. The mean±S.D. of the correlation coefficients between the two profiles was 0.95±0.06.
Identification of functional pathways
To analyze gene expression changes in the context of known biological pathways, we used PathWay Enrichment Analysis (PWEA)8 and identified 14 KEGG pathways that were deregulated following both Igf1 and Pacap treatments. These pathways can be organized in three main functional categories: cellular processes (focal adhesion; phagosome; antigen processing and presentation), metabolism (glycine, serine and threonine metabolism; glutathione metabolism; sfingolipid metabolism) and signal transduction (chemokine signaling; neuroactive ligand–receptor interaction; olfactory transduction; calcium signaling; GABAergic synapse; hedgehog signaling; TGF-beta signaling; MAPK signaling). All differently expressed pathways are shown in Supplementary Materials (Supplementary Figures S1A and S1M). Genes with significant deregulated changes in all these pathways are summarized in figure 2.

A comprehensive picture showing gene expression changes and crosstalk of pathways associated with Igf1 and Pacap rescue of apoptotic CGNs. This figure, designed using the MetaCore software (Thomson Reuters, New York, NY, USA), illustrates the gene expression profiles and the 14 signaling pathways that are involved in Igf1 and Pacap neuroprotection of apoptotic CGNs. Each encoded protein is labeled with two thermometers that indicate gene expression changes under the following experimental conditions: K5+Igf1 (first thermometer) and K5+Pacap (second thermometer). More specifically, downward thermometers have a blue color and reflect downregulated expression, whereas upward thermometers have a red color and reflect upregulated expression. Further explanation of symbols used in the pathway map are listed in Supplementary Figure S2.
Discussion
In recent years, the advent of full-genome sequencing and high-throughput technologies has offered a new approach to decode the mechanisms underlying neuronal apoptosis and survival, unraveling a systems biology-based perspective. The ability of a cell to promote or evade apoptosis depends on the activity of an integrated network of genes and their encoded proteins, which never work alone but interact with each other in highly structured and incredibly complex ways, forming molecular circuits that correspond perfectly to cell functional specifications.
This study reports for the first time a whole-genome analysis of neurons induced to apoptosis and rescued by growth factors. We found striking similarities in CGNs rescued by Igf1 and Pacap, despite the fact that they act on different receptors. This raised our interest in exploring individual genes and crosstalk of transcriptional pathways that govern CGN survival. In the following paragraphs, we discuss these transcriptional changes highlighting statistically significant changes in genes and functional pathways that accompany CGN rescue from neuronal apoptosis.
Cellular processes
Focal adhesions are important cell–matrix junctions that have an essential role in intracellular survival signaling. Our analysis revealed that the overexpression of genes encoding focal adhesion proteins during CGN rescue may favor cell survival. Their encoded proteins participate in the structural link between membrane receptors and the actin cytoskeleton (Tnc, Itgb1, Actn1, Actn4 and Actin cytoskeletal groups), whereas others are adaptors (Vav2, Bcar1, Src, Pik3r1 and Pdk1) that contribute to initiate downstream signaling events, leading to neuroprotection9–17 (Supplementary Figure S1A).
Growing evidence supports an active role for the dysregulated autophagy-lysosome pathway in neuronal cell death and neurodegeneration. Our microarray analysis showed that Igf1 and Pacap rescue is associated with the differential expression of autophagic regulator transcripts. Increased expressions of M6pr and Coro1a, as well as a decrease in Fcgr2a, have already been associated with anti-apoptotic effects.18–20 Similarly, a high level of Dync1li2 was found to have a key role in recycling endosome localization21 (Supplementary Figure S1B).
Differential regulation of genes encoding proteins involved in the antigen processing and presentation pathway may underlie preselection of immunologically important antigenic determinants in dying cells. Consistent with this view, we observed the differential expression of RT1-A1, Hspa5, Nf-Yb and Psme3 in rescued CGNs, all of which are known to exert an anti-apoptotic effect22–25 (Supplementary Figure S1C).
Metabolism
The sequence of biochemical changes occurring during amino-acid biosynthesis and metabolism in neuronal cells is fundamental in maintaining life-sustaining chemical transformations. In agreement with this is the differential expression of a number of genes encoding enzymes involved in the glycine, serine and threonine metabolism. Downregulation of Agtx, Psph, Pipox, which are mostly found in the peroxisomes, may represent an adaptive response to counteract stress-induced death signaling.26–28 In addition, a high level of Shmt2 has been found to confer neuroprotection29 (Supplementary Figure S1D).
Our analysis also revealed an overexpression of five genes encoding glutathione-dependent enzymes (Idh2, Gsta3, Gstm2, Gstm7 and Odc1) that have already been related to antioxidant and anti-apoptotic effects in various cell types, such as CGNs30–34 (Supplementary Figure S1D).
The sphingolipid pathway is also regulated during Igf1 and Pacap rescue and includes a significantly decreased expression of apoptotic Cers235 (Supplementary Figure S1D).
Signal transduction
A large number of genes mediating signaling cascades from the cell surface to the nucleus was involved in CGN rescue. Among these, we observed the differential expression of genes encoding chemokines, neuroactive ligands, their receptors or downstream enzymes (Cxcl12, Cx3cl1, Cxcl3, Gal, Adora1, F2rl3, Pthr1, Oxtr, mGluR4, Grin3b, Adcy6, Itk and PKCzeta) that are known to counteract cell death because of various harmful conditions36–48 (Supplementary Figures S1 E and F).
The reduction of a vast number of olfactory receptor transcripts, during apoptosis rescue, suggests that they may perform other functions than those previously reported49 (Supplementary Figure S1G).
Regulation of calcium homeostasis has been extensively involved in cell survival.50 Accordingly, we observed the overexpression of Cacna1d, Cacna1s, Slc8a1 and the decrease of P2rx6 and Ryr1, which are all known to control calcium signaling and render cells less vulnerable to a wide variety of apoptotic stimuli51–55 (Supplementary Figure S1H).
Our results also emphasized the critical role of GABAergic synaptic signaling during CGN rescue, as the overexpression of a sodium-dependent amino-acid transporter, Slc38a2, together with the above-described proteins (Adcy6, Cacna1d and Prkcg), is known to elicit survival responses56 (Supplementary Figure S1I).
The overexpression of three wnt ligands (Wnt4, Wnt5b and Wnt7a) is in line with the survival effects exerted by the Wnt family in neuronal cells.57 Moreover, the low expression of Ptch1 (the receptor for hedgehog) and Sufu (a common regulator of wnt and hedgehog signaling) in rescued CGNs is consistent with previous evidence obtained in different cell types58,59 (Supplementary Figure S1L).
A number of genes encoding TGF-beta signaling proteins are regulated by Igf1 and Pacap. Among these, high levels of Rps6kb1 and a loss of Smad6 have been demonstrated to counteract apoptosis60 (Supplementary Figure S1L).
One of the major molecular mechanisms for controlling cell survival is the MAPK signaling.61 In rescued CGNs, a number of genes encoding ligands or receptors related to the MAPK signaling (Pdgfb, Fgf21, Ntrk1 and Tnfrsf1a) have been involved in neuronal survival.62–65 Similarly, genes encoding enzymes, such as increased p90Rsk and Dusp5, as well as decreased Prkcg and Map4k1, have been associated with neuroprotection.66–69 We also observed the enhanced expression of genes encoding pro-survival heat shock proteins (Hspa1a, Hspa1b and Hspa2), genotoxic stress proteins (Gadd45a and Gadd45g) and transcription factors (Nr4a1, Jun, Fos and Srf)70–77 (Supplementary Figure S1M).
Conclusions
Our analysis offers for the first time a genomic view of the changes underlying the rescue from apoptosis by Igf1 and Pacap treatments of rat CGNs, the most common neuronal paradigm used to examine programmed cell death by trophic deprivation. Although acting through different upstream signaling pathways, most of the survival effects of Igf1 and Pacap were propagated by common transcriptional cascades, and showed striking similarities. These unpredicted results suggest the existence of a previously unknown transcriptional program specifying neuronal survival. Although commitment to apoptosis by CGNs is known to require de novo gene expression5 and has been the purpose of previous investigations, the transcriptional program underlying apoptosis rescue has been neglected. To our knowledge, this is the first time that investigated and compared transcriptional signaling pathways induced by rescue factors showed striking similarities. Elucidating the molecular mechanisms underlying neuronal apoptosis and rescue may contribute to the understanding of the molecular basis of neurodegeneration and aid the development of new therapeutic interventions. Several proteins encoded by differentially expressed genes, at the intersection of apoptosis and survival, are targets of pharmacological compounds (Supplementary Table S1). Their exploitation may help interfering with the intracellular pathways described here and guide novel therapeutic strategies for neurodegenerative diseases.
Materials and Methods
Materials
All the substances were obtained from Sigma-Aldrich (Milano, Italy), unless otherwise specified.
Neuronal cultures
Primary cultures of CGNs were prepared from 8-day-old Wistar rats (Charles River, Calco, Italy) and were cultured as previously described.7,78 In brief, cerebella were sliced and tissue was dissociated through trypsinization in 0.025% trypsin solution (15 min at 37 °C) and trituration in presence of DNase (0.01%) and trypsin inhibitor (0.05%). Dissociated cells were collected through centrifugation and resuspended in basal Eagle’s medium supplemented with 10% fetal calf serum, 25 mm KCl, 2 mm glutamine and 100 μg/ml gentamycin. Cytosine arabinofuranoside (10 μM) was added after 18 h of culture to inhibit the growth of non-neuronal cells.
Assessment of neurotrophic activity
After 6 days ‘in vitro’, extracellular KCl was shifted from 25 to 5 mm for neuronal apoptotic death induction. After two washes with serum-free BME containing 5 mm KCl, neurons were incubated with the same medium up to 48 h (K5), whereas control neurons were incubated with serum-free medium supplemented with 25 mm KCl (K25). K5 neurons were also treated with human recombinant Igf1 or Pacap.
The rescue effects of Igf1 and Pacap were determined by assessing neuronal viability, oligonucleosome formation and DNA fragmentation. Neuronal viability was assessed by counting the number of intact nuclei according to the method previously described.79 Culture medium was removed and replaced by 0.5 ml of a detergent containing lysing solution (0.5% ethylhexadecyldimethylammonium bromide, 0.28% acetic acid, 0.5% Triton X-100, 3 mm NaCl, 2 mM MgCl2, in PBS pH 7.4). After a few minutes, the cells were collected and intact viable nuclei were counted in a hemocytometer. Broken or damaged nuclei were not included in the counts. Quantitative determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) was performed by a photometric enzyme immunoassay (Cell Death Detection ELISAPLUS, Roche Diagnostics, Monza, Italy), as suggested by the manufacturer's protocol. Analysis of DNA fragmentation was performed with the Apoptotic DNA Ladder Kit (Roche Diagnostics), as specified in the instruction manual. Fragmented DNA (250 ng) was evaluated using a DNA 7500 chip and a 2100 Bioanalyzer (Agilent Technologies) with the protocol outlined by the manufacturer.
Microarray experiments
Microarray experiments were performed in serum-deprived cells after 6 h switch from 25 to 5 mm with or without treatments with 100 nm Pacap or 3.26 pm Igf1. Control cells were grown in serum-free medium supplemented with 25 mm KCl (K25). Total RNA was extracted with Trizol (Life Technologies, Monza, Italy) from four biological replicates for each of the experimental conditions (K25, K5, K5+Igf1, K5+Pacap). RNA integrity was confirmed by using a RNA chip and a 2100 Bioanalyzer (Agilent Technologies), with the protocol outlined by the manufacturer. Complementary RNAs (cRNAs) labeled with Cy3-CTP were synthesized from 1 μg of total RNA of each sample using the Low RNA Input Fluorescent Linear Amplification Kit (Agilent Technologies), following the manufacturer's protocol.
Aliquots (750 ng) of Cy3-labeled cRNA targets were hybridized on Whole Rat Genome Oligo Microarrays (Agilent Technologies). Microarray hybridization and washing were performed using reagents and instruments (hybridization chambers and rotating oven) as indicated by the manufacturer. Microarrays were scanned at 5-μm resolution using a GenePix Personal 4100 A microarray scanner and the GenePix Pro 6.0 acquisition and data-extraction software (Molecular Devices, Sunnyvale, CA, USA). Raw data were processed and analyzed with GeneSpringGX 13 (Agilent Technologies). To remove unreliable data, all genes from all samples were quality-filtered to include only probe data fulfiling all of the following criteria in all replicates of at least one out of four experimental conditions: the spot had <3% of saturated pixels at 532 nm; the spot was not flagged ‘bad’, ‘not found’ or ‘absent’; the spot was detectable well above background (signal-to-noise ratios at 532 nm >10). Filtering data by quality-control criteria short-listed 29 892 probes as our complete data set, out of a total of 41 012 probes present on the microarray. The microarray data have been deposited in the Gene Expression Omnibus (GEO) database under accession no. GSE67788.
Genes in our quality-filtered data set were screened by a one-way ANOVA using Welch's t-test, followed by the Benjamini and Hochberg false discovery rate (FDR) procedure as a multiple testing correction and the Tukey’s post hoc test. Genes with a corrected P-value<0.05 were selected as differentially expressed genes.
Pathway analysis
To analyze gene expression changes in the context of biological pathways, we used PWEA,8 a method to identify functionally related modules of genes, such as KEGG pathways, that correlate with phenotypic differences. PWEA, as implemented in VisANT mining system (http://visant.bu.edu/), differs from other enrichment methods in that it takes account of the functional correlations between genes in the module. This results in higher sensitivity with no loss in specificity.8 A PWEA analysis of the four experimental conditions (K25, K5, K5+Igf1 and K5+Pacap) was performed for two pair-wise comparisons (K5+Igf1 and K5+Pacap versus K5) using the quality-filtered data set. Overall, 245 rattus norvegicus pathways downloaded from KEGG were tested, excluding disease pathways, which are largely uninformative. Statistical significance was estimated from a background distribution generated by 5000 iterations of a permutation test, and the Benjamini–Hochberg FDR procedure using a P-value<0.05 as cutoff.
Real-time quantitative RT-PCR
To validate gene expression profiles observed by microarray technology, deregulated genes following Igf1 and Pacap treatments were re-evaluated using real-time quantitative PCR (RT-PCR) as previously described80 (Supplementary Table S2).
References
Arends MJ, Wyllie AH . Apoptosis: mechanisms and roles in pathology. Int Rev Exp Pathol 1991; 32: 223–254.
Oppenheim RW . Cell death during development of the nervous system. Annu Rev Neurosci 1991; 14: 453–501.
Mattson MP . Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid Redox Signal 2006; 8: 1997–2006.
Contestabile A . Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro . Cerebellum 2002; 1: 41–55.
D'Mello SR, Galli C, Ciotti T, Calissano P . Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 1993; 90: 10989–10993.
Cavallaro S, D'Agata V, Alessi E, Coffa S, Alkon DL, Manickam P et al. Gene expression profiles of apoptotic neurons. Genomics 2004; 84: 485–496.
Cavallaro S, Copani A, D'Agata V, Musco S, Petralia S, Ventra C et al. Pituitary adenylate cyclase activating polypeptide prevents apoptosis in cultured cerebellar granule neurons. Mol Pharmacol 1996; 50: 60–66.
Hung JH, Whitfield TW, Yang TH, Hu Z, Weng Z, DeLisi C . Identification of functional modules that correlate with phenotypic difference: the influence of network topology. Genome Biol 2010; 11: R23.
Naghdi P, Tiraihi T, Ganji F, Darabi S, Taheri T, Kazemi H . Survival, proliferation and differentiation enhancement of neural stem cells cultured in three-dimensional polyethylene glycol-RGD hydrogel with tenascin. J Tissue Eng Regen Med 2014.
Carlson TR, Hu H, Braren R, Kim YH, Wang RA . Cell-autonomous requirement for beta1 integrin in endothelial cell adhesion, migration and survival during angiogenesis in mice. Development 2008; 135: 2193–2202.
Quick Q, Skalli O . Alpha-actinin 1 and alpha-actinin 4: contrasting roles in the survival, motility, and RhoA signaling of astrocytoma cells. Exp Cell Res 2010; 316: 1137–1147.
Bunnell TM, Ervasti JM . Delayed embryonic development and impaired cell growth and survival in Actg1 null mice. Cytoskeleton 2010; 67: 564–572.
Vigorito E, Gambardella L, Colucci F, McAdam S, Turner M . Vav proteins regulate peripheral B-cell survival. Blood 2005; 106: 2391–2398.
Gonsior C, Biname F, Fruhbeis C, Bauer NM, Hoch-Kraft P, Luhmann HJ et al. Oligodendroglial p130Cas is a target of Fyn kinase involved in process formation, cell migration and survival. PLoS One 2014; 9: e89423.
Encinas M, Tansey MG, Tsui-Pierchala BA, Comella JX, Milbrandt J, Johnson EM Jr . c-Src is required for glial cell line-derived neurotrophic factor (GDNF) family ligand-mediated neuronal survival via a phosphatidylinositol-3 kinase (PI-3K)-dependent pathway. J Neurosci 2001; 21: 1464–1472.
Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B . Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc Natl Acad Sci USA 2010; 107: 11381–11386.
Park J, Kwon K, Kim SH, Yi MH, Zhang E, Kong G et al. Astrocytic phosphorylation of PDK1 on Tyr9 following an excitotoxic lesion in the mouse hippocampus. Brain Res 2013; 1533: 37–43.
Kanamori S, Waguri S, Shibata M, Isahara K, Ohsawa Y, Konishi A et al. Overexpression of cation-dependent mannose 6-phosphate receptor prevents cell death induced by serum deprivation in PC12 cells. Biochem Biophys Res Commun 1998; 251: 204–208.
Suo D, Park J, Harrington AW, Zweifel LS, Mihalas S, Deppmann CD . Coronin-1 is a neurotrophin endosomal effector that is required for developmental competition for survival. Nat Neurosci 2014; 17: 36–45.
Nguyen TH, Havari E, McLaren R, Zhang M, Jiang Y, Madden SL et al. Alemtuzumab induction of intracellular signaling and apoptosis in malignant B lymphocytes. Leuk Lymphoma 2012; 53: 699–709.
Palmer KJ, Hughes H, Stephens DJ . Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. MoL Biol Cell 2009; 20: 2885–2899.
Backman-Petersson E, Butcher GW, Hedlund G . Self-MHC class Ia (RT1-A(n)) protects cells co-expressing the activatory allogeneic MHC class Ib molecule (RT1-E(u)) from NK lysis. Int Immunol 2000; 12: 843–850.
Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS . Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem 2003; 278: 20915–20924.
Benatti P, Basile V, Merico D, Fantoni LI, Tagliafico E, Imbriano C . A balance between NF-Y and p53 governs the pro- and anti-apoptotic transcriptional response. Nucleic Acids Res 2008; 36: 1415–1428.
Sanchez N, Gallagher M, Lao N, Gallagher C, Clarke C, Doolan P et al. MiR-7 triggers cell cycle arrest at the G1/S transition by targeting multiple genes including Skp2 and Psme3. PLoS One 2013; 8: e65671.
Gamelin L, Capitain O, Morel A, Dumont A, Traore S, Anne le B et al. Predictive factors of oxaliplatin neurotoxicity: the involvement of the oxalate outcome pathway. Clin Cancer Res 2007; 13: 6359–6368.
Heese K, Nagai Y, Sawada T . Induction of rat L-phosphoserine phosphatase by amyloid-beta (1-42) is inhibited by interleukin-11. Neurosci Lett 2000; 288: 37–40.
Schrader M, Fahimi HD . Peroxisomes and oxidative stress. Biochim Biophys Acta 2006; 1763: 1755–1766.
Yoshida E, Atkinson TG, Chakravarthy B . Neuroprotective gene expression profiles in ischemic cortical cultures preconditioned with IGF-1 or bFGF. Brain Res Mol Brain Res 2004; 131: 33–50.
Kil IS, Shin SW, Yeo HS, Lee YS, Park JW . Mitochondrial NADP+-dependent isocitrate dehydrogenase protects cadmium-induced apoptosis. Mol Pharmacol 2006; 70: 1053–1061.
Mazurek B, Amarjargal N, Haupt H, Fuchs J, Olze H, Machulik A et al. Expression of genes implicated in oxidative stress in the cochlea of newborn rats. Hear Res 2011; 277: 54–60.
Pool-Zobel BL, Selvaraju V, Sauer J, Kautenburger T, Kiefer J, Richter KK et al. Butyrate may enhance toxicological defence in primary, adenoma and tumor human colon cells by favourably modulating expression of glutathione S-transferases genes, an approach in nutrigenomics. Carcinogenesis 2005; 26: 1064–1076.
Bogdani M, Henschel AM, Kansra S, Fuller JM, Geoffrey R, Jia S et al. Geoffrey R, Jia S, et al. Biobreeding rat islets exhibit reduced antioxidative defense and N-acetyl cysteine treatment delays type 1 diabetes. J Endocrinol 2013; 216: 111–123.
Sparapani M, Virgili M, Bardi G, Tregnago M, Monti B, Bentivogli M et al. Ornithine decarboxylase activity during development of cerebellar granule neurons. J Neurochem 1998; 71: 1898–1904.
Decraene C, Brugg B, Ruberg M, Eveno E, Matingou C, Tahi F et al. Identification of genes involved in ceramide-dependent neuronal apoptosis using cDNA arrays. Genome Biol 2002; 3: RESEARCH0042.
Cook A, Hippensteel R, Shimizu S, Nicolai J, Fatatis A, Meucci O . Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J Biol Chem 2010; 285: 10563–10571.
Hao F, Zhang NN, Zhang DM, Bai HY, Piao H, Yuan B et al. Chemokine fractalkine attenuates overactivation and apoptosis of BV-2 microglial cells induced by extracellular ATP. Neurochem Res 2013; 38: 1002–1012.
Lee YS, Won KJ, Park SW, Lee HW, Kim B, Kim JH et al. Mesenchymal stem cells regulate the proliferation of T cells via the growth-related oncogene/CXC chemokine receptor, CXCR2. Cell Immunol 2012; 279: 1–11.
Cordero-Llana O, Rinaldi F, Brennan PA, Wynick D, Caldwell MA . Galanin promotes neuronal differentiation from neural progenitor cells in vitro and contributes to the generation of new olfactory neurons in the adult mouse brain. Exp Neurol 2014; 256: 93–104.
Fatokun AA, Stone TW, Smith RA . Adenosine receptor ligands protect against a combination of apoptotic and necrotic cell death in cerebellar granule neurons. Exp Brain Res 2008; 186: 151–160.
Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE . Inhibiting protease-activated receptor 4 limits myocardial ischemia/reperfusion injury in rat hearts by unmasking adenosine signaling. J Pharmacol Exp Ther 2008; 324: 1045–1054.
Ono T, Inokuchi K, Ogura A, Ikawa Y, Kudo Y, Kawashima S . Activity-dependent expression of parathyroid hormone-related protein (PTHrP) in rat cerebellar granule neurons. Requirement of PTHrP for the activity-dependent survival of granule neurons. J Biol Chem 1997; 272: 14404–14411.
Ceanga M, Spataru A, Zagrean AM . Oxytocin is neuroprotective against oxygen-glucose deprivation and reoxygenation in immature hippocampal cultures. Neurosci Lett 2010; 477: 15–18.
Borodezt K, D'Mello SR . Decreased expression of the metabotropic glutamate receptor-4 gene is associated with neuronal apoptosis. J Neurosci Res 1998; 53: 531–541.
Fukumori R, Takarada T, Nakamichi N, Kambe Y, Kawagoe H, Nakazato R et al. Requirement of both NR3A and NR3B subunits for dominant negative properties on Ca2+ mobilization mediated by acquired N-methyl-D-aspartate receptor channels into mitochondria. Neurochem Int 2010; 57: 730–737.
Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation 2006; 114: 388–396.
Kelly E, Won A, Refaeli Y, Van Parijs L . IL-2 and related cytokines can promote T cell survival by activating AKT. J Immunol 2002; 168: 597–603.
Crisanti P, Leon A, Lim DM, Omri B . Aspirin prevention of NMDA-induced neuronal death by direct protein kinase Czeta inhibition. J Neurochem 2005; 93: 1587–1593.
Watt WC, Sakano H, Lee ZY, Reusch JE, Trinh K, Storm DR . Odorant stimulation enhances survival of olfactory sensory neurons via MAPK and CREB. Neuron 2004; 41: 955–967.
Zundorf G, Reiser G . Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal 2011; 14: 1275–1288.
Gao L, Blair LA, Salinas GD, Needleman LA, Marshall J . Insulin-like growth factor-1 modulation of CaV1.3 calcium channels depends on Ca2+ release from IP3-sensitive stores and calcium/calmodulin kinase II phosphorylation of the alpha1 subunit EF hand. J Neurosci 2006; 26: 6259–6268.
Antony C, Mehto S, Tiwari BK, Singh Y, Natarajan K . Regulation of L-type voltage gated calcium channel CACNA1S in macrophages upon Mycobacterium tuberculosis infection. PLoS One 2015; 10: e0124263.
Cross JL, Boulos S, Shepherd KL, Craig AJ, Lee S, Bakker AJ et al. High level over-expression of different NCX isoforms in HEK293 cell lines and primary neuronal cultures is protective following oxygen glucose deprivation. Neurosci Res 2012; 73: 191–198.
Slater M, Barden JA, Murphy CR . The purinergic calcium channels P2X1,2,5,7 are down-regulated while P2X3,4,6 are up-regulated during apoptosis in the ageing rat prostate. Histochem J 2000; 32: 571–580.
Suzuki M, Nagai Y, Wada K, Koike T . Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochem Biophys Res Commun 2012; 429: 18–23.
Nishimura T, Sai Y, Fujii J, Muta M, Iizasa H, Tomi M et al. Roles of TauT and system A in cytoprotection of rat syncytiotrophoblast cell line exposed to hypertonic stress. Placenta 2010; 31: 1003–1009.
Marchetti B, L'Episcopo F, Morale MC, Tirolo C, Testa N, Caniglia S et al. Uncovering novel actors in astrocyte-neuron crosstalk in Parkinson's disease: the Wnt/beta-catenin signaling cascade as the common final pathway for neuroprotection and self-repair. Eur J Neurosci 2013; 37: 1550–1563.
Chen XL, Chinchilla P, Fombonne J, Ho L, Guix C, Keen JH et al. Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol Cell Biol 2014; 34: 3855–3866.
Lee DY, Deng Z, Wang CH, Yang BB . MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci USA 2007; 104: 20350–20355.
Bakhshayesh M, Zaker F, Hashemi M, Katebi M, Solaimani M . TGF- beta1-mediated apoptosis associated with SMAD-dependent mitochondrial Bcl-2 expression. Clin Lymphoma Myeloma Leuk 2012; 12: 138–143.
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME . Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999; 286: 1358–1362.
Tseng HC, Dichter MA . Platelet-derived growth factor-BB pretreatment attenuates excitotoxic death in cultured hippocampal neurons. Neurobiol Dis 2005; 19: 77–83.
Leng Y, Wang Z, Tsai LK, Leeds P, Fessler EB, Wang J et al. FGF-21, a novel metabolic regulator, has a robust neuroprotective role and is markedly elevated in neurons by mood stabilizers. Mol Psychiatry 2015; 20: 215–223.
Marlin MC, Li G . Biogenesis and function of the NGF/TrkA signaling endosome. Int Rev Cell Mol Biol 2015; 314: 239–257.
Taoufik E, Petit E, Divoux D, Tseveleki V, Mengozzi M, Roberts ML et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc Natl Avad Sci USA 2008; 105: 6185–6190.
Liu Y, Li J, Yang J, Ji F, Bu X, Zhang N et al. Inhibition of PKCgamma membrane translocation mediated morphine preconditioning-induced neuroprotection against oxygen-glucose deprivation in the hippocampus slices of mice. Neurosci Lett 2008; 444: 87–91.
Li B, Yi P, Zhang B, Xu C, Liu Q, Pi Z et al. Differences in endoplasmic reticulum stress signalling kinetics determine cell survival outcome through activation of MKP-1. Cell Signal 2011; 23: 35–45.
Stankiewicz TR, Ramaswami SA, Bouchard RJ, Aktories K, Linseman DA . Neuronal apoptosis induced by selective inhibition of Rac GTPase versus global suppression of Rho family GTPases is mediated by alterations in distinct mitogen-activated protein kinase signaling cascades. J Biol Chem 2015; 290: 9363–9376.
Mengozzi M, Cervellini I, Villa P, Erbayraktar Z, Gokmen N, Yilmaz O et al. Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc Natl Avad Sci USA 2012; 109: 9617–9622.
Lowenstein DH, Chan PH, Miles MF . The stress protein response in cultured neurons: characterization and evidence for a protective role in excitotoxicity. Neuron 1991; 7: 1053–1060.
Park HK, Cho AR, Lee SC, Ban JY . MPTP-induced model of Parkinson's disease in heat shock protein 70.1 knockout mice. Mol Med Rep 2012; 5: 1465–1468.
Daugaard M, Jaattela M, Rohde M . Hsp70-2 is required for tumor cell growth and survival. Cell Cycle 2005; 4: 877–880.
Lin CR, Yang CH, Huang CE, Wu CH, Chen YS, Sheen-Chen SM et al. GADD45A protects against cell death in dorsal root ganglion neurons following peripheral nerve injury. J Neurosci Res 2011; 89: 689–699.
Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C et al. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet 2009; 5: e1000604.
Courtney MJ, Akerman KE, Coffey ET . Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci 1997; 17: 4201–4211.
Tong L, Perez-Polo R . Brain-derived neurotrophic factor (BDNF) protects cultured rat cerebellar granule neurons against glucose deprivation-induced apoptosis. J Neural Transm 1998; 105: 905–914.
Chang SH, Poser S, Xia Z . A novel role for serum response factor in neuronal survival. J Neurosci 2004; 24: 2277–2285.
Levi G, Aloisi F, Ciotti MT, Gallo V . Autoradiographic localization and depolarization-induced release of acidic amino acids in differentiating cerebellar granule cell cultures. Brain Res 1984; 290: 77–86.
Volonte C, Ciotti MT, Battistini L . Development of a method for measuring cell number: application to CNS primary neuronal cultures. Cytometry 1994; 17: 274–276.
Maino B, Ciotti MT, Calissano P, Cavallaro S . Transcriptional analysis of apoptotic cerebellar granule neurons following rescue by gastric inhibitory polypeptide. Int J Mol Sci 2014; 15: 5596–5622.
Acknowledgements
We gratefully acknowledge Maria Patrizia D’Angelo, Alfia Corsino and Cristina Calì for their administrative support and Francesco Marino for technical expertise. CD and Y-CC acknowledge support from the National Institutes of Health grant R01GM103502-05.
Data and material availability
Microarray expression data files can be obtained from the NIH GEO (http://www.ncbi.nlm.nih.gov/geo/).
Author information
Authors and Affiliations
Contributions
BM and VD carried out RT-PCR analysis and drafted the manuscript; CS, MTC and AC performed neuronal culture experiments, Y-CC and CD performed pathway enrichment analysis, PC and SC conceived and designed the study. SC completed microarray analysis and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Cell Death Discovery website (http://www.nature.com/cddiscovery)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Maino, B., D’Agata, V., Severini, C. et al. Igf1 and Pacap rescue cerebellar granule neurons from apoptosis via a common transcriptional program. Cell Death Discovery 1, 15029 (2015). https://doi.org/10.1038/cddiscovery.2015.29
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/cddiscovery.2015.29
This article is cited by
-
Transcriptional landscapes at the intersection of neuronal apoptosis and substance P-induced survival: exploring pathways and drug targets
Cell Death Discovery (2016)
-
Cracking the code of neuronal apoptosis and survival
Cell Death & Disease (2015)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}