
Abstract
In previous experiments that simulated conditions on primordial volcanic islands, we demonstrated the abiotic formation of hydrophobic porphyrins. The present study focused on the question whether such porphyrins can be metalated by prebiotically plausible metal ion sources. We used water-insoluble octaethylporphyrin (H2oep) as a model compound. Experiments were conducted in a nitrogen atmosphere under cyclic wet–dry conditions in order to simulate the fluctuating environment in prebiotic rock pools. Wetting–drying proved to be a crucial factor. Significant yields of the metalloporphyrins (20–78% with respect to H2oep) were obtained from the soluble salts MCl2 (M = Mg, Fe, Co, Ni and Cu) in freshwater. Even almost insoluble minerals and rocks metalated the porphyrin. Basalt (an iron source, 11% yield), synthetic jaipurite (CoS, 33%) and synthetic covellite (CuS, 57%) were most efficient. Basalt, magnetite and FeCl2 gave considerably higher yields in artificial seawater than in freshwater. From iron sources, the highest yields, however, were obtained in an acidic medium (hydrochloric acid with an initial pH of 2.1). Under these conditions, iron meteorites also metalated the porphyrin. Acidic conditions were considered because they are known to occur during eruptions on volcanic islands. Octaethylporphyrinatomagnesium(II) did not form in acidic medium and was unstable towards dissolved Fe2+. It is therefore questionable whether magnesium porphyrins, i.e. possible ancestors of chlorophyll, could have accumulated in primordial rock pools. However, abiotically formed ancestors of the modern cofactors heme (Fe), B12 (Co), and F430 (Ni) may have been available to hypothetical protometabolisms and early organisms.
Similar content being viewed by others

Introduction
In order to perform their function, many proteins―particularly those involved in group transfer and redox reactions (including electron transfer)―require small or medium-sized organic molecules as “coenzymes” or, more generally, “cofactors.” The porphyrin-type cofactors (PTCs) are metal complexes that consist of a macrocyclic tetrapyrrole ligand and a tightly bound metal ion, such as Fe2+/3+, Mg2+, Co3+, and Ni2+ (Kaim et al. 2013). They ubiquitously occur in all kingdoms of life. Here we use the term “porphyrin-type” in a wider sense to include, for example, metal complexes of contracted porphyrins (e.g., coenzyme B12) and partially reduced ones (e.g., cofactor F430). Heme and chlorophylls are the best known representatives of the PTCs. Heme-containing cytochrome P450 enzymes are thought to have existed for more than 3.5 billion years (Nelson et al. 1993). Thus, the PTC family is evolutionary very old and could well have a prebiotic origin.
An abiotic formation of a PTC ancestor under early Earth conditions would probably have proceeded in two steps: first, the synthesis of the porphyrin and, subsequently, the incorporation of the metal ion. At least one alternative pathway is conceivable: an open-chain tetrapyrrole could have first bonded to the metal ion and then cyclized to form the macrocyclic tetrapyrrole complex. Examples for this kind of reaction are known (Wood and Thompson 2007). However, they require conditions that are of little or no prebiotic relevance (e.g., organic solvents, bromo substituents). Therefore, the first pathway appears to be more plausible. Indeed, we have already experimentally demonstrated its initial step, the formation of porphyrins under reasonable prebiotic conditions (Fox and Strasdeit 2013).
The prebiotic scenario for our porphyrin synthesis is based on the chemical and physical environments on primordial volcanic islands. The synthesis consists of four steps: (i) embedding of amino acids in sea salt crusts near coastal lava flows; (ii) thermal transformation of these amino acids into pyrroles, accompanied by release of hydrochloric acid from the sea salt; (iii) condensation of the (volatile) pyrroles and hydrochloric acid in cooler rock pools; (iv) hydrochloric acid-catalyzed condensation of the pyrroles with formaldehyde and oxidation by nitrite to give conjugated oligopyrroles, including porphyrins. The entire sequence was experimentally simulated in the laboratory. Moreover, the starting materials involved (sea salt, amino acids, formaldehyde, nitrite) are generally regarded as “prebiotic.” The overall yield was low but must be seen against the facts that (i) the alkyl-substituted porphyrins formed were water insoluble and (ii) porphyrins are extremely stable under geological conditions (Callot and Ocampo 2000). Therefore, the alkylporphyrins must have accumulated over time.
In related studies, Lindsey’s group has demonstrated the non-enzymatic synthesis of porphyrinogens from α-aminoketones and β-diketones or β-ketoesters in water (Lindsey et al. 2009, 2011; Soares et al. 2012a, b; Taniguchi et al. 2012; Soares et al. 2013a). A spontaneous prebiotic formation of the starting materials used is, to our knowledge, unknown. Therefore, this reaction cannot be regarded as a “primordial-soup synthesis” (as defined in: Strasdeit and Fox 2013). However, early cell ancestors (Pohorille 2009) may have produced the starting materials in primitive protometabolisms. Already half a century ago, the formation of porphyrins from pyrroles and aldehydes has been recognized as a potentially prebiotic reaction (see for example: Szutka 1964). In line with this idea, the reaction of 3,4-dialkylpyrroles with formaldehyde in aqueous micellar solution has recently been studied (Alexy et al. 2015). Despite low (probably prebiotically realistic) pyrrole concentrations, good yields were obtained.
Early metal incorporation experiments using pyrrole, paraformaldehyde, and metal salts under simulated prebiotic conditions gave only tiny yields of metal porphyrins (Hodgson and Baker 1967). In contrast, the reaction of Mn2+, Co2+ and Ni2+ salts with the hydrophilic uroporphyrins in dilute aqueous solutions (pH range 6.3–9.1, 37 °C) resulted in 40 to 84% yields of the respective metal complexes (Frydman and Stevens 1968). In a more recent study, modifications and extensions of this uroporphyrin–metal salt approach were described, including the use of additional metal ions (Mg2+, Fe2+, Cu2+, Zn2+, and Pd2+; Soares et al. 2013b).
However, it still remains unclear how hydrophobic porphyrins such as octaethylporphyrin (H2oep, Fig. 1) could have been able to bind metal ions under prebiotic conditions. Hydrophobic alkylporphyrins are products of the above-described amino acid route (Fox and Strasdeit 2013) and also form from appropriately substituted aminoketones and diketones (Soares et al. 2013a). In addition, prebiotic hydrophilic or amphiphilic porphyrins could have been transformed into more hydrophobic ones by thermal decarboxylation, for example in volcanic environments. Recently, solvent-free syntheses of metal(II) porphyrin complexes have been accomplished by reacting solid metal ion sources (magnesium oxide and hydrated metal acetates, respectively) with solid meso-tetraphenylporphyrin (Schneider et al. 2016; Ralphs et al. 2017). The results suggest that solid–solid reactions should be considered as routes to metal porphyrins on the early Earth, even though the reported syntheses were not performed under simulated prebiotic conditions.

Structures of (a) 2,3,7,8,12,13,17,18-octaethylporphyrin (H2oep), (b) 2,3,7,8,12,13,17,18-octaethylporphyrinato metal(II) complexes ([M(oep)]), (c) H2oepO (an oxygenation product of H2oep), and (d) [Fe(oepO–H)] (an iron complex of the oxygenated porphyrin)
Obviously, the water insolubility of alkylporphyrins poses a problem for metal complex formation in aqueous media. Moreover, some metals relevant to PTC ancestors had rather low concentrations in the Hadean–early Archean ocean, namely iron, cobalt, and nickel, but not magnesium (Holland 1984; Saito et al. 2003). The iron concentration, for example, is often reported to have been ~0.1 mmol L−1 (see for example: Holland 1973; Saito et al. 2003; Canfield 2005; Li et al. 2013). In addition to dissolved metal salts, several minerals could have served as metal ion sources (Hazen 2013), but here again their virtual insolubility seems to be problematic. Our idea was that the reaction between an alkylporphyrin and a (soluble or insoluble) metal ion source may nevertheless be possible in wet–dry cycles. It is presumed that wet–dry cycles produced a fluctuating environment in prebiotic rock pools (Lathe 2004; Deamer 2014). Therefore, alternating wetting and drying has been used in several prebiotic simulation experiments, with a major focus on oligomer formation (see for example: Lahav et al. 1978; Lahav and White 1980; Saetia et al. 1993; Olasagasti et al. 2011; Mamajanov et al. 2014; Forsythe et al. 2015; Rodriguez-Garcia et al. 2015). Recently, we have studied glycine oligomerization and other test reactions in a newly developed automated wet–dry apparatus, which can be operated under strictly anaerobic conditions (Fox et al. 2018).
During the late Hadean–early Archean period, the alternation between wet and dry phases must have been particularly pronounced on the shores of active volcanic islands. At that time, the Earth had a considerably faster rotation rate (Walker 1982; Walker et al. 1983; Zahnle and Walker 1987; Lathe 2004; Varga et al. 2006). The day length 3.9 Ga ago has been estimated between ~4 and ~17 h; 14 h (i.e., tides every ~7 h) appears to be a good provisional value for use in simulation experiments (Lathe 2006). Thus, around the time when life emerged, wet–dry cycling in rock pools was probably nearly twice as fast as today.
During the dry phase of a wet–dry cycle, solid–solid surface contacts may allow insoluble substances, such as alkylporphyrins and minerals, to react with each other. In the subsequent wet phase, the solids are separated again, so that in the next dry phase fresh surfaces can come into contact and react. We have tested this idea with octaethylporphyrin and several prebiotically plausible iron and some copper, cobalt, nickel, and magnesium sources, and the results are reported here.
Materials and Methods
Chemicals, Minerals, Rocks, and Meteorites
2,3,7,8,12,13,17,18-Octaethylporphyrin (H2oep, 97%) and chlorido(octaethylporphyrinato)iron(III) ([FeCl(oep)], 97%) were purchased from TriPorTech, Selmsdorf, Germany. Octaethylporphyrinatocobalt(II) ([Co(oep)], 98%) and octaethylporphyrinatonickel(II) ([Ni(oep)], 98%) were obtained from PorphyChem, Dijon, France. Octaethylporphyrinatocopper(II) ([Cu(oep)], 97%), ferrozine (≥97%), and neocuproine (99%) were from Sigma-Aldrich. Octaethylporphyrinatomagnesium(II) ([Mg(oep)], Johnson et al. 1980), octaethylporphyrinatoiron(II) ([Fe(oep)], Konarev et al. 2009), μ-oxo-bis[octaethylporphyrinatoiron(III)] ([{Fe(oep)}2(μ-O)], Dolphin et al. 1978), octaethylporphyrinium dichloride ((H4oep)Cl2, Ogoshi et al. 1973), cobalt(II) sulfide, and nickel(II) sulfide (Glemser and Schwarzmann 1981) were prepared by literature procedures. The following compounds were used in high-purity form: ammonium iron(II) sulfate hexahydrate (99.997%, Sigma-Aldrich), iron powder (99.998%, Alfa Aesar, particle size ≤6 μm), iron(II) chloride tetrahydrate (99.99%, Sigma-Aldrich), iron(II,III) oxide (99.995%, Sigma-Aldrich), and iron(III) oxide (99.999%, Sigma-Aldrich). All other metal salts and all organic solvents were of analytical grade. Double distilled water, which was prepared in a quartz glass distillation apparatus (BD 50, Westdeutsche Quarzschmelze), was used throughout the study.
Well-formed crystals of pyrrhotite (Nikolaevskiy mine, Dalnegorsk, Russia) were purchased from Wilfried Kittler, Freiberg, Germany. Pyrite (Mererani, Tanzania), olivine (Shigar, Pakistan) and chalcopyrite (Alban, France) samples were obtained from Jentsch Mineralien, Extertal, Germany. X-ray powder diffraction showed that the chalcopyrite also contained pyrite. The ankerite sample (Oust, Ariège, France) was also from Jentsch Mineralien. Its Fe content was 2.8%, its carbonate-C content 11.8% (determined by Mikroanalytisches Labor Pascher, Remagen, Germany). Magnetite crystals (Bahia, Brazil) were obtained from Mineraliengrosshandel Hausen, Telfs, Austria. For all minerals used, including the synthetic ones, the phase purity was checked by X-ray powder diffraction.
The basalt used in this study was a product of the 2010 eruption of the Piton de la Fournaise volcano on the island of La Réunion, Indian Ocean. The samples were collected in 2011. They were taken from the interior of larger basalt chunks in order to minimize biological and weathering contamination. Within two weeks after collection, the material was dried and sterilized at 150 °C for 24 h and then stored in a closed container at ~0 °C. Major elements (determined by X-ray fluorescence spectrometry at the Landesamt für Geologie, Rohstoffe und Bergbau, Freiburg, Germany), expressed as oxides, were: SiO2 45.36, Al2O3 13.91, CaO 12.83, FeO 8.02, MgO 7.05, Fe2O3 4.27, Na2O 3.02, TiO2 2.77, K2O 0.82, P2O5 0.32, and MnO 0.18%. The SiO2, MgO and (Na2O + K2O) contents together define this rock as a basalt (Gill 2010). The komatiite used in this study was a relatively fresh one from the 3.27 Ga Weltevreden Formation, Barberton Greenstone Belt, South Africa (sample SA 564–1; Kareem 2005; Puchtel et al. 2013). Our sample contained 0.9% Fe2+ (determined by Mikroanalytisches Labor Pascher); for further analytical data, see Puchtel et al. (2013). The sample was provided by Dr. Igor Puchtel of the University of Maryland, College Park, USA.
Prior to use, all minerals and rocks were crushed and passed through a 500-μm sieve. The fraction with particle sizes <500 μm was used for the experiments, unless otherwise stated.
Shavings of the iron meteorites Shişr 043 and Twannberg II were used in this study. The major constituents of Shişr 043 are iron (91.00%) and nickel (8.07%), with the remainder being mostly cobalt and phosphorus (Al-Kathiri et al. 2006). The size of its shavings was ~0.5 × 0.25 mm. Twannberg II has a higher iron content (95.5%) and contains ~4.5% nickel (Hofmann et al. 2009). The Twannberg II shavings used were ~5 × 1 mm in size. Both meteorite samples were provided by Dr. Beda Hofmann of the Naturhistorische Museum Bern, Switzerland. Prior to use, the samples were treated with hydrochloric acid (2 mol L−1) for 10 min and then thoroughly washed with water.
Artificial seawater was prepared by dissolving 41.20 g (705 mmol) of sodium chloride, 1.12 g (15 mmol) of potassium chloride, 16.26 g (80 mmol) of magnesium chloride hexahydrate, and 2.21 g (15 mmol) of calcium chloride dihydrat in water and making up to 1 L. The reasons why this composition was chosen as a model for primordial sea salt have been discussed elsewhere (Fox and Strasdeit 2013). The resulting overall salt content of 5.0% was higher than that of modern seawater (3.5%). This is in accordance with the idea that the ocean in the Archean was more saline than today (Knauth 1998, 2005).
Reactions in the Wet–Dry Apparatus (WDA)
The experimental setup and operating principle of the WDA have been described in detail elsewhere (Fox et al. 2018). In brief, the apparatus consists of a Schlenk flask, a heat-insulated glass riser, a reflux condenser, a 50-mL reservoir, and a time controlled magnetic valve. The aqueous mixture in the flask is heated with an oil bath, causing the water to slowly evaporate. During this wet phase, the water vapor rises through the riser into the reflux condenser. From the condenser, the water flows into the reservoir and collects there. When the water has completely evaporated from the flask, the residue is dry heated. At the end of the dry phase, the valve opens for a short time and allows the water to flow back from the reservoir into the flask. At this point, the next wet phase starts. Before and during an experiment, the WDA can be purged with nitrogen or another suitable gas. The apparatus is automated. Key experimental parameters (for example, the durations of the wet and dry phases) can be varied.
Typical procedure: Special care was taken to exclude oxygen during the whole experiment. To a Schlenk flask containing 30 mL of deoxygenated water, artificial seawater or hydrochloric acid (10 mmol L−1), 1.00 mg (1.87 μmol) of octaethylporphyrin and a defined amount of an iron source were added against a countercurrent of nitrogen gas (99.999% purity). Iron was always present in excess. The flask was attached to the rest of the WDA, and the whole system was purged with nitrogen for 60 h. Then the wet–dry cycling was started by heating the flask to 150 °C, which resulted in the evaporation of the water. After 7 h, the magnetic valve opened for 15 min so that the water flowed back into the hot flask. Thus, the reaction mixture was rehydrated, and the next cycle started. The mixture was subjected to 13 consecutive wet–dry cycles. After the last dry phase, the flask was transferred for analysis into a glove box containing an argon atmosphere (~1 ppm O2). The same procedure was applied with sources of metals other than iron. In these cases, only water and typically 1 mmol of the respective metal source were used.
Analytical Instrumentation
High-Performance Thin-Layer Chromatography (HPTLC)
Silica gel 60 HPTLC plates (10 × 20 cm, MS grade, Merck) were used as the stationary phase. Bands of the sample and standard solutions were applied by an Automatic TLC Sampler 4 from Camag (Muttenz, Switzerland). Typically, 18 bands with a length of 6 mm each were applied on a single plate, and the plates were developed up to a migration distance of 60 mm in a flat bottom chamber at ambient temperature and humidity. After development, the plates were dried at ambient conditions and scanned at a single wavelength with a TLC Scanner 3 (Camag) in the absorption mode. Additionally, absorption spectra of the bands were recorded in the 200–700 nm range with the TLC Scanner 3 to check the correct substance assignment and the purity. The colors of the bands were visible with the naked eye.
LDI–TOF/TOF Mass Spectrometry
Mass spectra were measured with an Autoflex III spectrometer from Bruker Daltonics (Bremen, Germany). A toluene solution of coronene, phthalocyanine, and the fullerenes C60 and C70 was used for external calibration of the instrument. Typically, 2 μL of a solution of the analyte in dichloromethane were transferred to a polished steel target plate and dried at room temperature. Sample preparation was conducted under an argon atmosphere in a glove box (~1 ppm O2).
UV–Visible Spectroscopy
Absorption spectra were recorded on a Specord 210 spectrometer from Analytik Jena (Jena, Germany). The scan speed was 1 nm s−1, and the resolution was 0.5 nm. Dichloromethane solutions of the analytes were measured in the wavelength range 250–800 nm in a gas-tight quartz glass cuvette under anaerobic conditions, unless otherwise stated. For the colorimetric iron determination with ferrozine, the spectrometer was operated in the single wavelength mode at 562 nm. Disposable polystyrene cuvettes were used which were discarded after each measurement to avoid cross-contamination.
Atomic Force Microscopy (AFM) on Magnetite Surfaces
AFM was performed using a JPK NanoWizard II system (JPK Instruments, Berlin, Germany), which was placed on a vibration insulated table. The images were acquired in tapping mode with a PPP-NCH silicon cantilever (Nanosensors, Neuchatel, Switzerland). The drive frequency was ~300 kHz, and the scan rate was between 0.8 and 1.0 Hz. All measurements were performed in air. The free software Gwyddion (version 2.50; Nečas and Klapetek 2012) was used for image processing.
The samples were natural octahedral magnetite crystals (~6 mm edge length, 500–600 mg). Prior to use, the crystal surfaces were manually wet polished with progressively finer silicon carbide abrasive paper (grit sizes P400, P600, P800, P1000, P1200 and P2000) and rinsed repeatedly with water. After drying in a desiccator, the polished crystals were stored in a pure argon atmosphere to prevent oxidation of the surfaces. A few crystals were crushed using tungsten carbide equipment, and the phase purity of the obtained powder was checked by X-ray diffraction. Before a wet–dry experiment was started, the polished magnetite surfaces were examined by AFM. Then the crystal was exposed to 12 wet–dry cycles using 30 mL of deaerated water. All experiments were carried out in a nitrogen atmosphere. After the experiments the samples were stored under an argon atmosphere for no longer than three days before they were examined by AFM. At least ten randomly chosen areas of each magnetite sample were analyzed.
Fluorescence Microscopy on H2oep Deposited on Magnetite
Fluorescence microscopy was performed using an Axioscope 2 microscope equipped with EC Epiplan-Neofluar 20x and 40x objectives, filter set 09 (BP 450–490 exciter filter, FT 510 beam splitter, LP 515 emission filter), and HBO 100 illuminator (Carl Zeiss Microscopy GmbH, Jena, Germany). Photomicrographs were taken with a 10.6 megapixel CMOS camera SC100 (Olympus, Hamburg, Germany) attached to the microscope. The camera was controlled by cellSens Standard V1.15 software including Manual Process Control, which allowed focus stacking and thus the production of high depth of field images. The magnetite samples were manually polished natural octahedral crystals as used for AFM. In each experiment, one magnetite crystal and 5 mg of H2oep were exposed to wet–dry cycles in 30 mL of deaerated water in a nitrogen atmosphere. The magnetite crystal was recovered at the end of a dry phase, and the H2oep on its surfaces was analyzed by fluorescence microscopy. The high-energy edge of the Q IV band of H2oep was within the excitation range used (450–490 nm).
X-Ray Powder Diffractometry
Powder diffraction data were obtained on a D8 Focus diffractometer from Bruker AXS (Karlsruhe, Germany) using CuKα radiation (λ = 1.5418 Å). The instrument was equipped with a Sol-X energy dispersive detector. Diffractograms were measured in the 2θ range 5–90°.
Analytical Procedures
Extraction of the Metalloporphyrins and Sample Preparation
The dry residue obtained from the reaction with an iron source was transferred into a glove box (argon atmosphere, ~1 ppm O2) where it was extracted with deaerated dichloromethane eleven times (once with 2 mL and ten times with 1 mL). For the mass spectrometric and absorption spectroscopic measurements, 4 mL were taken from the extract. The remaining 8 mL were stirred in air with 2 mol L−1 hydrochloric acid for 1 h. Under these conditions, all possibly formed iron porphyrin complexes were converted to [FeCl(oep)] (Maricondi et al. 1969; Dolphin et al. 1976; Goff 1981). Then the organic phase was separated, and the solvent was removed under reduced pressure. The residue was dissolved in 1 mL of dichloromethane. Aliquots of this solution were used for the yield determination by HPTLC. The extraction procedure was tested with various known amounts of [Fe(oep)]. It was found that no demetalation occurred and the complex was completely converted to [FeCl(oep)] under these conditions.
When artificial seawater or water-soluble iron sources were used, a slightly modified procedure was applied. First, the salt crust which had formed in the experiment was extracted with a total of 12 mL of deaerated dichloromethane. In this case only 200 μL of the extract were taken for mass spectrometric and absorption spectroscopic measurements. Then the salt crust was dissolved in 20 mL of 2 mol L−1 hydrochloric acid, 7 mL of dichloromethane were added, and the mixture was stirred for 10 min. Next, the resulting two phases (aqueous and organic) were combined with the first dichloromethane extract, and the combined mixture was stirred in air for 1 h. The subsequent steps were carried out as described above.
Residues obtained from reactions with sources of metals other than iron were processed in the same manner, except that the treatment with hydrochloric acid was omitted.
Determination of the Metalloporphyrin Yields by High-Performance Thin-Layer Chromatography
For the quantitative determination of iron porphyrin complexes, sample and standard solutions in dichloromethane were applied to HPTLC plates. [FeCl(oep)] served as the standard. The plates were developed in a saturated chamber with a mixture of n-hexane, methanol and chloroform (8:2:1, v/v/v). Densitometry was performed by absorption measurement at 386 nm. UV–visible spectra revealed that during chromatography [FeCl(oep)] was transformed into the μ-oxido dimer [{Fe(oep)}2(μ-O)]. This behavior is known from chromatography on alumina (Ivashin et al. 1996).
In order to check the HPTLC results, the iron content of selected samples was additionally determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES; Mikroanalytisches Labor Pascher). Prior to analysis, each dichloromethane extract (the same as used for HPTLC) was filtered through a 0.45 μm PTFE membrane to remove particulate material. An aliquot of the filtrate was used for ICP-AES after evaporation of the solvent at ambient temperature and pressure. HPTLC measured only the iron in the iron porphyrin, while with ICP-AES the whole iron content of the sample was determined. As can be seen in Table 1, the ICP-AES values are close to, but systematically higher than, the HPTLC values. Probably our efforts to remove the particulate material were not entirely successful, and the ICP-AES data contained a contribution from non-porphyrin-bound iron. However, the ICP-AES values still support the reliability of the HPTLC results, as they are only 3 to 8 percentage points higher.
The porphyrin complexes of other metals were also quantified by HPTLC. The basic procedure was the same as described for [FeCl(oep)], but with different mobile phases and detection wavelengths. A mixture of n-hexane and toluene (1:1, v/v) was used for [Cu(oep)] and [Ni(oep)], and a mixture of dichloromethane and methanol (100:2, v/v) for [Co(oep)]. In all three cases, the detection was at 398 nm. The yield of [Mg(oep)] was determined with toluene as the mobile phase and detection at 572 nm.
As the metal ion sources were used in excess of the porphyrin, the yields given in Tables 1 and 3 were calculated with respect to H2oep.
Quantitative Determination of Dissolved Iron by the Ferrozine Method
Samples were prepared by suspending mineral or rock particles in either water, artificial seawater or 10 mmol L−1 hydrochloric acid. Typically after 60 h, the sample was centrifuged. The supernatant was used to determine the iron that had been liberated from the solid. A 500-μL aliquot was diluted either with 500 μL of 20 mmol L−1 hydrochloric acid (in case of water or artificial seawater as solvent) or with 500 μL of 10 mmol L−1 hydrochloric acid (in case of 10 mmol L−1 hydrochloric acid as solvent). The method used for the subsequent iron determination was adopted from Riemer et al. (2004). To prepare the iron-detection reagent, 16 mg (32.5 μmol) of ferrozine, 7 mg (33 μmol) of neocuproine, 964 mg (12.5 mmol) of ammonium acetate, and 881 mg (5.0 mmol) of ascorbic acid were dissolved in 5 mL of water. Neocuproine was used to mask otherwise disturbing copper ions. To 300 μL of an aqueous sample solution, 300 μL of 10 mmol L−1 hydrochloric acid and 100 μl of the iron-detection reagent were added. The solution was thoroughly mixed and after 30 min transferred into a disposable cuvette. The absorbance at 562 nm was measured against a reagent blank consisting of 600 μL of hydrochloric acid and 100 μL of the iron-detection reagent. The iron content was calculated using a calibration curve, which was obtained from ammonium iron(II) sulfate hexahydrate standard solutions. The standard solutions were treated in the same manner as the actual samples. If necessary, samples were diluted with double distilled water prior to use.
Results and Discussion
Background of the Wet–Dry Simulation Experiments
The aim of this study was to simulate organic–inorganic interactions in rock pools, especially in tide-washed ones, on primordial volcanic islands. The experiments were performed in a newly developed wet–dry apparatus (WDA, see Materials and Methods). The temperature, pH, duration of the wet–dry cycles, content material of a rock pool, and an oxygen-free atmosphere could be simulated in the WDA.
It is reasonable to assume that two essentially different types of rock pools existed at primordial coasts. One type was close to the shoreline. It was affected by the tides and therefore contained seawater (tide pools). The other type was more remote from the shoreline and occasionally filled with rainwater (freshwater). In such freshwater rock pools, the wet–dry cycles were much more irregular because rainfall did not occur as periodic as tides. Nevertheless, we performed the freshwater and salt-water experiments with the same wet–dry frequency to ensure comparability of the results. In addition to rain, water vapor from the interaction of seawater with hot lava represents another freshwater source. Sea salt-containing aerosols or mixtures of seawater and rain water can form brackish water in rock pools. If lava is sufficiently hot, it releases hydrochloric acid (HCl) from seawater. This has been observed, for example, at the coasts of Hawai’i and La Réunion (Edmonds and Gerlach 2006; Bhugwant et al. 2009). The HCl originates from the decompostion of solid MgCl2 hydrates (Wiberg 2001). It is plausible that this process already occurred at primordial volcanic coasts because Earth’s early ocean was probably highly saline (Knauth 1998, 2005; Izawa et al. 2010). Therefore, Hadean–early Archean rock pools could also have contained acidic water. Based on these considerations, we performed our experiments with freshwater, acidic freshwater, and saltwater as reaction media.
In this study, the hydrophobic octaethylporphyrin (H2oep, Fig. 1) was used as a model compound. The reasons for this are explained in the Introduction. If porphyrins were present in primordial rock pools, they inevitably came into contact with minerals and rocks. Since rock pools at volcanic coasts are depressions in solidified lava, our experiments focussed on igneous rocks and their mineral components. However, other minerals that were probably present on the early Earth (Hazen 2013) were also included in the series of experiments reported here.
Metalation of Octaethylporphyrin by Iron Sources
General Remarks
Besides igneous rocks (basalt and komatiite), we studied the volcanism-related minerals magnetite and olivine as iron sources. In addition, other plausible Hadean minerals such as ankerite, pyrite, and pyrrhotite, as well as the soluble salt iron(II) chloride were used. Large amounts of metallic iron could have been delivered to the early Earth by meteorites, especially during the late heavy bombardment. Therefore, we also employed synthetic iron powder and shavings of the iron meteorites Twannberg II and Shişr 043.
After each wet–dry experiment, the iron porphyrin complexes that had formed were completely transformed into the air-stable [FeCl(oep)]. This ensured a reliable quantification of the complexed iron. Experimental and analytical details are given in Materials and Methods.
Spectroscopic Results and Oxidative Processes
Figure 2a shows, as an example, the LDI–TOF/TOF mass spectrum of the dichloromethane-soluble products from a wet–dry experiment with H2oep and basalt in artificial seawater. The three main peak groups correspond to (i) unreacted H2oep ([H2oep + H]+ at m/z 535.3, overlapped by H2oep+; compare Fig. 2d), (ii) [Fe(oep)]+ (m/z 588.3), and (iii) [FeCl(oep)]+ (m/z 623.3). Mass spectra of the standard [FeCl(oep)] usually showed the corresponding molecular peak at m/z 623.3 (Fig. 2c). However, under LDI conditions the chlorido ligand was partly or, at higher laser energies, completely lost, resulting in a signal at m/z 588.3 ([Fe(oep)]+). Not unexpectedly, the same m/z value occurred in the spectrum of the iron(II) complex [Fe(oep)] (Fig. 2b). Consequently, the formation of [Fe(oep)] in chloride-containing media cannot be proved by LDI–TOF/TOF MS, because the m/z 588.3 signal may also be due to [FeCl(oep)].

LDI–TOF/TOF mass spectra: dichloromethane extract of the dry residue from a simulation experiment with H2oep and basalt in artificial seawater (a); standard substances: [Fe(oep)] (b), [FeCl(oep)] (c), H2oep (d), and [Cu(oep)] (e). Calculated values are given in parentheses
Oxidation of Fe(II) to Fe(III) may already occur in the wet–dry cycles. Thus, even in the absence of chloride, the m/z 588.3 signal does not necessarily indicate the presence of the Fe(II) complex [Fe(oep)] in the sample. Avoiding oxidation in wet–dry experiment is technically very demanding (Fox et al. 2018). This is especially true when highly oxygen-sensitive species such as [Fe(oep)] and Fe(II)(aq) are present in low amounts. Under these conditions, low residual oxygen concentrations in the gas phase are sufficient to cause oxidation. Therefore, we cannot rule out the possibility that Fe(II) was partly oxidized to Fe(III) in our wet–dry experiments. Fe(III) also reacts with H2oep to form a complex. This was confirmed through wet–dry experiments with iron(III) oxide as the iron source.
In order to test whether [Fe(oep)] was the final product of wet–dry experiments with Fe(II) sources, the dichloromethane extract from a suitable experiment (iron(II) chloride and H2oep in hydrochloric acid) was deliberately oxidized by air. The UV–visible spectra before and after oxidation differed significantly (Fig. 3a and d, respectively). The spectrum of the oxidized extract was virtually identical to that of an equimolar mixture of [FeCl(oep)] and H2oep (Fig. 3e), demonstrating that [FeCl(oep)] had formed and H2oep was present. If [FeCl(oep)] was already the major iron species in the untreated extract, exposure to air would not have caused considerable changes in the UV–visible spectrum, because this complex is not oxidizable. Thus, the occurrence of spectral changes indicates that originally an Fe(II) complex, very probably [Fe(oep)], was present. In line with this, the spectrum of the untreated extract showed a shoulder at 388 nm, which is the position of the Soret band of [Fe(oep)] (compare Fig. 3a and b). The Q-bands of [Fe(oep)] could not be unambiguously identified, because of overlap with bands of H2oep and (H4oep)Cl2 (see next paragraph).

UV–visible spectra in dichloromethane: extract of the dry residue from a simulation experiment with FeCl2 and H2oep under acidic conditions (a), [Fe(oep)] (b), octaethylporphyrinium dichloride (H4oep)Cl2 (c), extract of the residue after air oxidation (d), equimolar mixture of [FeCl(oep)] and H2oep (e), [FeCl(oep)] (f), and H2oep (g)
The strong bands at 404 and ~422 nm in the spectrum of the non-oxidized extract (Fig. 3a) coincide with the Soret band of H2oep (Fig. 3g) and (H4oep)Cl2 (Fig. 3c), respectively. The formation of (H4oep)Cl2 from H2oep and hydrochloric acid has been described in the literature (Ogoshi et al. 1973). In contrast to the insoluble H2oep, (H4oep)Cl2 is slightly soluble in acidic water. Because of this, (H4oep)Cl2 may be at least partly responsible for the comparatively high yields observed in wet–dry experiments under acidic conditions (Table 1). (H4oep)Cl2 also played an important role in the oxidation of the dichloromethane extract (see above), whereby [Fe(oep)] was oxidized to [FeCl(oep)] (reaction 1). The formation of the chlorido complex was to some extent unexpected because air oxidation usually leads to the oxido dimer [{Fe(oep)}2(μ-O)] (James 1978). (H4oep)Cl2, however, provided not only the chloride ions needed as ligands but also protons to trap the oxide ion (see the formal reactions 2 and 3). Free HCl―a potential alternative source of chloride and protons―was not available in the dichloromethane extract, because the extraction was performed on a dry residue.
In the spectrum of the oxidized extract (Fig. 3d), the Soret band at 402 nm and the Q-bands at 503, 506, 538, 571, 599 and 626 nm could be unambiguously assigned to H2oep. The shoulder at ~382 nm was consistent with the presence of [FeCl(oep)], which has its Soret band at 380 nm (Fig. 3f). Assignment of the Q-bands of [FeCl(oep)], however, was difficult, because in the relevant region H2oep dominated the spectrum. Only the broad band at ~640 nm could be identified as a Q-band of [FeCl(oep)]. In the reference spectrum, it occurred at 636 nm and was absent in the spectrum of H2oep. As mentioned above, the simultaneous presence of [FeCl(oep)] and H2oep in the oxidized extract was demonstrated by comparison with the spectrum of an equimolar mixture of the two compounds (Fig. 3d and e). The 1:1 mixture did not only reproduce the band positions but also the relative band intensities, indicating a metal complex yield of around 50% in the wet–dry experiment. This is in good agreement with the actually measured yield of 58% (entry 1c in Table 1).
The mineral pyrite turned out to be different from the other iron sources. It is known that when pyrite comes into contact with oxygen-free water, hydrogen peroxide (H2O2) is formed (Borda et al. 2001). H2O2 can oxygenate a carbon atom at one of the meso positions of H2oep or [Fe(oep)], resulting in the formation of H2oepO and [Fe(oepO–H)], respectively (Fig. 1; Balch 2000, Kalish et al. 2000, 2001). Indeed, LDI–TOF/TOF spectra of the extracts from our wet–dry experiments with H2oep and pyrite or chalcopyrite showed the signals of H2oepO (m/z found 550.4, calcd 550.37) and [Fe(oepO–H)] (m/z found 603.3, calcd 603.28). The observation that chalcopyrite also led to the formation of the oxygenated species is plausibly explained by its pyrite content (see Materials and Methods). These results indicate that alkylporphyrin complexes analogous to [Fe(oepO–H)] may have formed by oxidation in a prebiotic water–pyrite environment. [Fe(oepO–H)] can be regarded as the Fe(III) complex of triply deprotonated H2oepO, at least in one of its resonance structures.
The Influence of Low pH
Strongly acidic conditions―for example, rain of pH ˂ 2―occur on present-day volcanic islands as a result of eruptions (Staudacher et al. 2009; see also the Introduction). It therefore seems plausible that acidic environments also existed on primordial volcanic island. This led us to examine whether low pH influenced the formation of [Fe(oep)] in wet–dry experiments. Brønsted acidity is often an important factor in metal complex formation. On the one hand, metal ions can be mobilized from solids at low pH so that they become available for complex formation. On the other hand, metal ions and protons compete for ligands; therefore, complex formation with protonable ligands often becomes increasingly difficult with decreasing pH. In our experiments, the yields of the iron complex were always significantly higher under acidic than under neutral conditions (Table 1). For basalt, magnetite, metallic iron, and Twannberg II, for example, the yields were in the 40% range in acidic solution, compared with 11% or less without acid. Obviously, [Fe(oep)] was quite resistant to protonolysis (i.e., protonation of the coordinated porphyrinate), and positive effects of low pH on complex formation predominated.
Indeed, at least three effects can be identified which could have promoted the formation of [Fe(oep)] under acidic conditions:
-
(i)
At the beginning of a wet phase, the hydrochloric acid, which was used in the experiments to obtain acidic conditions, had a low concentration (~0.04%, corresponding to pH 2). However, hydrogen chloride and water form an azeotrope containing ~20% HCl. Therefore, the solutions became increasingly acidic as the evaporation progressed. As a result, H2oep became increasingly protonated to (H4oep)Cl2. As noted before, its water solubility strongly suggests that (H4oep)Cl2 was beneficial to the formation of the iron complex. This assumption was substantiated by experiments with FeCl2 as the iron source (see below).
-
(ii)
Furthermore, the protonation of H2oep could have facilitated the complex formation mechanistically. Indeed, one possible pathway of metalloporphyrin formation is the protonation of the porphyrin molecule on one side―which deforms the ring―followed by incorporation of the metal ion from the opposite side (Khosropour and Hambright 1972).
-
(iii)
The third effect of low pH is the solubilization of relatively large amounts of iron from most, but not all, iron sources used (Table 2). This corresponds well with higher yields obtained under acidic conditions (Table 1). For ankerite and komatiite, the yields in acidic medium were still low in absolute terms, which is consistent with the low release of iron from these sources. In the case of pyrrhotite (Fe1–xS), the yield was also low, despite the fact that considerable amounts of iron were released. The reason for this is unclear. At least we can say that metal sulfides do not generally give low yields, as we have shown with CuS, CoS and NiS (see below).
The importance of dissolved iron ions can be seen, for example, with magnetite. In nearly neutral solution, only a small amount of iron (<0.015 μmol) was released from this mineral within 60 h (Table 2). Consequently, only a low yield of the iron complex (1%) was obtained in the wet–dry experiment with magnetite and H2oep under neutral conditions (Table 1, entry 3a). In line with this, the soluble iron source FeCl2 gave much higher yields of 25% and 12% (with 1 mmol and 3 μmol, respectively, of FeCl2; Table 1, entries 1a and b). This latter result also showed that the yield increased with increasing concentration of iron ions. These experiments were performed in neutral solution. In acidic solution, the yield was nearly five times as large (58% as compared with 12%; Table 1, entry 1c), indicating that the protonation of the porphyrin, particularly the formation of the soluble (H4oep)Cl2, promoted the complex formation (see above).
In all but one of the experiments that started under acidic conditions, the pH remained low (between 2.2 and 5.3) until the end (Table 2). Only in the case of the carbonate mineral ankerite was the HCl solution neutralized from pH 2.1 to pH 7.2. At the beginning of the ankerite experiment, 0.3 mmol of H3O+ and 9.8 mmol of carbonate were present. Thus, only 1.5% of the total amount of carbonate were sufficient to neutralize the solution. As a consequence of the resulting low H3O+ concentration, no protonation of H2oep occurred. Moreover, the iron content in the solution over the mineral remained low. These factors explain why only a low yield of 2% was obtained from a wet–dry experiment with ankerite and H2oep despite the initial presence of hydrochloric acid (Table 1).
In addition to minerals and rocks, we investigated metallic iron and samples of two iron meteorites in wet–dry experiments. The reason was that large amounts of meteoritic iron must have been delivered to the Earth’s surface in the late Hadean–early Archean. The meteoritic materials used were shavings of Shişr 043 and Twannberg II. In order to remove surface impurities, such as rust, the shavings were briefly treated with concentrated hydrochloric acid and then thoroughly washed with water shortly before the experiments. As expected, metallic iron and the meteorite shavings released only small amounts of iron ions under neutral conditions (Table 2) and consequently the yields of the iron complex were poor (<0.5%, Table 1). Under acidic conditions, however, the iron metal was partly converted to soluble FeCl2 by redox reaction with HCl, and therefore the yields were much higher (metallic iron, 42%; Shişr 043, 29%; Twannberg II, 46%). The fact that Shişr 043 produced the lowest yield among the three iron sources can be explained by this meteorite’s relatively high nickel content (see Materials and Methods) which may have impeded the reaction with HCl. Indeed, the amount of iron released by acid from Shişr 043 was only half as large as for metallic iron and Twannberg II (Table 2). In summary, these results show that even unweathered iron meteorites can act as metal sources in the formation of iron porphyrins under cyclic wet–dry conditions, provided the reaction medium is sufficiently acidic.
The Role of Wet–Dry Cycling
A periodic increase in acidity increased the concentrations of dissolved iron and protonated H2oep, illustrating one of the particular effects of wet–dry cycling (see above). In order to identify additional effects, we performed experiments in neutral freshwater. First, basalt and magnetite were studied without wet–dry cycling and in the absence of H2oep. After 60 h, the supernatants had become slightly alkaline and only small amounts of iron ions had been released (<0.015 μmol, Table 2). Significantly more iron dissolved when wet–dry cycling was included (basalt, 0.21 μmol; magnetite, 0.07 μmol). In acidic medium, however, much larger amounts of iron dissolved, both with and without wet–dry cycles. This shows that wet–dry cycling facilitates the release of iron, but also that acid is more effective in this regard.
Another set of experiments was performed with H2oep and either FeCl2 or basalt, again under neutral conditions and without wet–dry cycling; the experimental duration was the same as for standard wet–dry experiments. In one type of experiment, after evaporation of the water, the reactants were dry heated. In another type, constant reflux was applied so that the reaction mixtures remained in the wet phase. In none of these experiments were significant amounts of iron porphyrins found (yields <1%). Under comparable wet–dry conditions, however, the yields obtained from FeCl2 and basalt were 12 and 11%, respectively (Table 1). From these results we conclude that wet–dry cycles are essential for the iron porphyrin complexes to be formed in appreciable yields in neutral medium.
To gain insight into the possible mechanical effects that wet–dry cycling may have on minerals, we studied polished magnetite surfaces in neutral water. In each experiment, only one crystal was used in order to avoid abrasion by mechanical interaction between crystals. Before and after the wet–dry experiment, the surfaces were examined by atomic force microscopy (Fig. 4). Marked morphological alterations were evident: the initially smooth polished surface showed a large increase in roughness after the experiment. Even the grinding marks, which resulted from the polishing procedure, were no longer visible. It is reasonable to assume that more iron ions can be released because of the greater roughness of the crystal surfaces and the additional surface area of the ablated material. This can explain why in neutral water the amount of dissolved iron was higher after wet–dry cycling.

AFM images of naturally grown magnetite single crystal surfaces. a Typical example of an untreated, polished magnetite surface. The red arrows and dashed lines indicate grinding marks resulting from the polishing procedure (see Materials and Methods). b Typical example of a magnetite surface after 12 wet–dry cycles, using water and a pure nitrogen atmosphere
We also studied how the mechanical contact between H2oep and magnetite changed with number of wet–dry cycles. A single magnetite crystal in neutral water was used. Figure 5a shows a fluorescence photomicrograph of H2oep on a surface of the magnetite crystal after one wet–dry cycle. The H2oep crystals still had sharp edges and were more or less equally distributed over the surface. While at this early stage some crystals were visible even with the naked eye, individual crystals were no longer apparent after 12 wet–dry cycles. Instead, H2oep appeared “smeared” over parts of the magnetite surface (Fig. 5b). Furthermore, a red-violet layer of H2oep was observed on the surface of the reaction flask. The crystals had probably been crushed by mechanical effects (see above), increasing the surface area of the porphyrin. In addition, it may be that the porphyrin patches were in closer contact with the mineral surface than the individual crystals were before. Both effects of the wet–dry cycling could have promoted the complex formation.

Fluorescence photomicrographs of H2oep on a polished magnetite single crystal surface. a After one wet–dry cycle at the end of the dry phase, b after 12 wet–dry cycles at the end of the last dry phase. Focus stacking was performed. Scale bar, 50 μm
Complex Formation in Saltwater
Primordial rock pools close to the shoreline must have contained saltwater (see Introduction). Therefore, we also performed experiments with artificial seawater as the reaction medium. Here the focus was on iron(II) chloride, basalt and magnetite as iron sources. The yields obtained were in the relatively narrow range of 19–24% (Table 1), which was higher than in the corresponding experiments with freshwater but lower than in the experiments under acidic conditions. Additional preliminary experiments did not give a clear picture of how exactly the salts influenced complex formation. Therefore, this point will not be discussed further.
In summary, our experimental results clearly indicate that iron porphyrins could have formed by wet–dry cycling in primordial rock pools, whereby the yield strongly depends on the conditions of the aqueous environment and the type of iron source. It is worthwhile to consider that sources of metals other than iron could also have reacted with porphyrins. A first hint of this possibility came from an experiment with basalt in which the formation of [Cu(oep)] was observed (Fig. 2a and e). Consequently, further experiments with selected copper, cobalt, nickel and magnesium ion sources were carried out. With the exception of copper, these metals occur in biological porphyrin-type cofactors (see Introduction).
Metalation of Octaethylporphyrin by Copper, Cobalt, Nickel and Magnesium Sources
Wet–dry cycling was performed under neutral conditions with the metal(II) chlorides and with synthetic forms of the minerals covellite (CuS), jaipurite (CoS), millerite (NiS) and brucite (Mg(OH)2). In all cases, the metal(II) complexes [M(oep)] (Fig. 1) were detected at the end of the experiment (Table 3), and their identities were confirmed by authentic standards. Yields were between 4 and 78%. The chlorides of Cu2+, Co2+, Ni2+ and Mg2+ are all readily soluble in water, while the minerals used have low or very low solubilities. This could at least partly explain why, for a given metal ion, the chloride always gave a higher yield than the mineral. The same was observed for iron sources in freshwater where the yields from FeCl2 were higher than from the minerals (Table 1). It should be noted that during a wet–dry cycle the metal(II) chlorides changed their hydration state and thus the form in which they could react with H2oep. In the wet phase, they were completely dissolved. During the subsequent transition from the wet to the dry phase, solid hydrates MCl2 ∙ n H2O crystallized, the value of n being dependent on the metal ion. Then, when the temperature of the residue increased towards its final value in the dry phase (150 °C), the hydrates could lose water of crystallization. Magnesium chloride, for example, first formed the hexahydrate and at 117 °C transformed into the tetrahydrate (Wiberg 2001).
Formation of the copper complex [Cu(oep)] was observed not only with CuCl2 and CuS but also with basalt. The mass spectrum of the products from a wet–dry experiment with basalt and H2oep in artificial seawater was dominated by the signals of [FeCl(oep)]+, [Fe(oep)]+, [H2oep + H]+ and H2oep+ (Fig. 2a). Another, smaller, signal at m/z 595.3 could be unambiguously assigned to [Cu(oep)]+ on the basis of its mass value (calcd 595.29), characteristic isotope pattern, and comparison with a standard (Fig. 2e). The fact that clearly detectable amounts of [Cu(oep)] formed despite the low copper content of the basalt (0.01%, compared with 6.2% Fe2+ and 3.0% Fe3+) could indicate an exceptionally high stability and/or an efficient formation mechanism of this complex. Also pointing in this direction is the observation that despite its extremely low solubility (Wiberg 2001), CuS gave a surprisingly high yield (57%) of [Cu(oep)]. Indeed, the “stability index” puts Cu2+, but not Fe2+, among the divalent metal ions that form the most stable octaalkylporphyrinato complexes; high stability together with facile complex formation can also account for the existence of copper geoporphyrins (Buchler 1975). The stability difference between Cu2+ and Fe2+, however, should not obscure the fact that Fe2+ also binds tightly to porphyrinates.
It is noteworthy that despite its formula, the compound CuS is not copper(II) sulfide. Based on its crystal structure (Evans Jr and Konnert 1976) and assuming a „normal“covalent situation, it may be assigned the formula \( {\left({\mathrm{Cu}}^{+}\right)}_2\left(\mathrm{C}{\mathrm{u}}^{2+}\right)\left({\mathrm{S}}_2^{2\hbox{--}}\right)\left({\mathrm{S}}^{2\hbox{--}}\right) \) (see for example: Wells 1984). However, certain physical properties and electronic structure calculations suggest that other formulas may be more appropriate, for example, \( {\left({\mathrm{Cu}}^{+}\right)}_3\left({\mathrm{S}}_2^{2\hbox{--}}\right)\left({\mathrm{S}}^{\hbox{--}}\right) \), \( {\left({\mathrm{Cu}}^{+}\right)}_3\left({\mathrm{S}}_2^{\hbox{--}}\right)\left({\mathrm{S}}^{2\hbox{--}}\right) \), \( {\left({\mathrm{Cu}}^{4/3+}\right)}_3\left({\mathrm{S}}_2^{2\hbox{--}}\right)\left({\mathrm{S}}^{2\hbox{--}}\right) \) or \( {\left({\mathrm{Cu}}^{3/2+}\right)}_2\left(\mathrm{C}{\mathrm{u}}^{+}\right)\left({\mathrm{S}}_2^{2\hbox{--}}\right)\left({\mathrm{S}}^{2\hbox{--}}\right) \) (Nozaki et al. 1991; Liang and Whangbo 1993; Mazin 2012; Kumar et al. 2013). The latter formulas imply that some kind of redox reaction must have been involved in the formation of the Cu(II) complex [Cu(oep)] from CuS and H2oep.
Theoretical calculations by Saito et al. (2003) showed that precipitation of highly insoluble copper sulfides probably caused an exceedingly low concentration of dissolved copper in the Archean ocean. Thus, the abiotic formation of copper complexes from dissolved copper ions would have been severely hampered. In this context, our observation that CuS can serve as an efficient copper ion source is of interest because it explains how porphyrinato copper(II) complexes may still have formed. CuS (covellite) is among the 420 minerals that were probably widely distributed and/or had a significant volume at or near the Hadean Earth’s surface (Hazen 2013). If copper porphyrins ever were functional components of (proto)metabolisms, they later lost their function(s). Today, the occurrence of copper porphyrins in organisms appears to be limited to feather pigments―turacin and turacoverdin (Dyck 1992)―for which no role in metabolism is known.
Cobalt, nickel and magnesium, in contrast to copper, are constituents of porphyrin-type cofactors that are involved in various metabolic processes (see Introduction). Our wet–dry experiments showed that prebiotic ancestors of these cofactors may have formed in rock pools in the late Hadean–early Archean. Relatively high yields of [Co(oep)] were obtained from CoCl2 (78%) and CoS (33%). The yields of [Ni(oep)] were lower (20% from NiCl2 and 6% from NiS) and similar to those of [Mg(oep)] (32% from MgCl2 and 4% from Mg(OH)2). In the case of [Mg(oep)], it turned out that neutral conditions were necessary for complex formation. This is consistent with the known fact that this complex is readily demetalated by traces of acid (Johnson et al. 1980). Remarkably, acid had the opposite effect on the formation of the corresponding Fe2+ complex, as discussed above. This difference is in line with the higher stability index of octaalkylporphyrinato iron(II) complexes (Buchler 1975), which also explains the following observations. When dissolved Mg2+ and Fe2+ ions were simultaneously present in a wet–dry experiment, formation of the iron complex clearly predominated and no [Mg(oep)] could be detected. In a further experiment in which pre-synthesized [Mg(oep)] was exposed to wet–dry cycles in the presence of FeCl2, transmetalation was observed, i.e. iron replaced magnesium in the porphyrinato ring. These results imply that in primordial rock pools, porphyrinato magnesium complexes could have existed only under non-acidic and essentially iron-free conditions. Therefore, abiotically formed chlorophyll precursors may have been less common than precursors of the heme group.
Summary and Conclusions
We have simulated the metalation of hydrophobic porphyrins in primordial rock pools by use of a wet–dry apparatus. Octaethylporphyrin (H2oep) was used as a model porphyrin. The main results of this study are:
-
(i)
In neutral freshwater, the salts MCl2 (M = Mg, Fe, Co, Ni and Cu) gave significant yields of the respective metalloporphyrins (20–78%), despite the fact that the porphyrin H2oep was completely water insoluble. Using FeCl2 as an example, we showed that wet–dry cycling was crucial. With only dry heating or refluxing, the yields were at least ten times lower.
-
(ii)
Surprisingly, under wet–dry conditions, even almost insoluble minerals and rocks could metalate H2oep in freshwater. The yields, however, were mostly low. Notable exceptions were the yields obtained with basalt as an iron source (11%), synthetic jaipurite (CoS, 33%) and synthetic covellite (CuS, 57%). Experiments with selected iron sources (basalt, magnetite and FeCl2) showed that yields were considerably higher in artificial seawater than in freshwater.
-
(iii)
The highest yields from iron sources were obtained in hydrochloric acid, which was used to simulate the acidic conditions that can result from eruptions on volcanic islands. Here again, wet–dry cycling was important: the initially low HCl concentration periodically increased during the evaporation phases because water and HCl form an azeotrope with a relatively high acid concentration (~20%). Separate experiments without the porphyrin showed that the minerals and rocks generally released much larger amounts of iron in acidic than in neutral medium. A further reason for the beneficial effect of hydrochloric acid is the protonation of the insoluble H2oep to form the slightly soluble (H4oep)Cl2.
-
(iv)
Prebiotic porphyrinato magnesium complexes are interesting as potential primitive versions of chlorophyll. We have, however, demonstrated that their formation on primordial volcanic islands was probably much more limited than the formation of the corresponding iron complexes. For example, octaethylporphyrinatomagnesium(II) did not form under acidic conditions, in sharp contrast to the iron complex whose formation was facilitated by acid (see above). In addition, we observed that octaethylporphyrinatomagnesium(II) was destroyed in a transmetalation reaction when dissolved Fe2+ was present.
-
(v)
Iron meteorites were also effective iron sources, albeit only under acidic conditions. We investigated two meteorites in wet–dry cycles and obtained yields of the porphyrinato iron complex of 29 and 46%.
-
(vi)
Based on our experimental results, it seems likely that on primordial volcanic islands, metalloporphyrins could have formed abiotically, even from insoluble or nearly insoluble porphyrins and metal ion sources. Porphyrinato complexes of iron, cobalt, nickel and copper, but perhaps not magnesium, may have accumulated in rock pools. If this is true, potential ancestors of porphyrin-type cofactors were available to hypothetical protometabolisms and early organisms and thus might have been involved in the origin and early evolution of life. In particular, abiotically formed iron porphyrins could have participated in electron transfer, which is one of the most basic biochemical processes. Our results are also consistent with the hypothesis that the porphyrin-type cofactors B12 and F430, which contain cobalt and nickel, respectively, are evolutionary very old (Fraústo da Silva and Williams 2001).
-
(vii)
The results of our experiments have implications for the use of metalloporphyrins as biosignatures. The abiotic metalation of porphyrins in wetting–drying processes may have occurred not only on Earth but also, for example, on early Mars. Therefore, should metalloporphyrins be discovered in Noachian or early Hesperian rocks on Mars, they could well be “false positive” biosignatures (Fox and Strasdeit 2017).
References
Alexy EJ, Hintz CW, Hughes HM, Taniguchi M, Lindsey JS (2015) Paley’s watchmaker analogy and prebiotic synthetic chemistry in surfactant assemblies. Formaldehyde scavenging by pyrroles leading to porphyrins as a case study. Org Biomol Chem 13:10025–10031
Al-Kathiri A, Hofmann BA, Gnos E, Eugster O, Welten KC, Krähenbühl U (2006) Shişr 043 (IIIAB medium octahedrite): the first iron meteorite from the Oman desert. Meteorit Planet Sci 41:A217–A230
Balch AL (2000) Coordination chemistry with meso-hydroxylated porphyrins (oxophlorins), intermediates in heme degradation. Coord Chem Rev 200–202:349–377
Bhugwant C, Siéja B, Bessafi M, Staudacher T, Ecormier J (2009) Atmospheric sulfur dioxide measurements during the 2005 and 2007 eruptions of the Piton de La Fournaise volcano: implications for human health and environmental changes. J Volcanol Geotherm Res 184:208–224
Borda MJ, Elsetinow AR, Schoonen MA, Strongin DR (2001) Pyrite-induced hydrogen peroxide formation as a driving force in the evolution of photosynthetic organisms on an early earth. Astrobiology 1:283–288
Buchler JW (1975) Static coordination chemistry of metalloporphyrins. In: Smith KM (ed) Porphyrins and metalloporphyrins. Elsevier, Amsterdam, pp 195–202
Callot HJ, Ocampo R (2000) Geochemistry of porphyrins. In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook, vol 1. Academic Press, San Diego, pp 349–398
Canfield DE (2005) The early history of atmospheric oxygen: homage to Robert M. Garrels. Annu Rev Earth Planet Sci 33:1–36
Deamer D (2014) The origin of life. In: Losos JB, Baum DA, Futuyma DJ, Hoekstra HE, Lenski RE, Moore AJ, Peichel CL, Schluter D, Whitlock MC (eds) The Princeton guide to evolution. Princeton University Press, Princeton, pp 120–126
Dolphin D, Sams JR, Tsin TB, Wong KL (1976) Synthesis and Mossbauer spectra of octaethylporphyrin ferrous complexes. J Am Chem Soc 98:6970–6975
Dolphin DH, Sams JR, Tsin TB, Wong KL (1978) Mössbauer–Zeeman spectra of some octaethylporphyrinato- and tetraphenylporphinatoiron(III) complexes. J Am Chem Soc 100:1711–1718
Dyck J (1992) Reflectance spectra of plumage areas colored by green feather pigments. Auk 109:293–301
Edmonds M, Gerlach TM (2006) The airborne lava–seawater interaction plume at Kīlauea volcano, Hawai’i. Earth Planet Sci Lett 244:83–96
Evans HT Jr, Konnert JA (1976) Crystal structure refinement of covellite. Am Mineral 61:996–1000
Forsythe JG, Yu S-S, Mamajanov I, Grover MA, Krishnamurthy R, Fernández FM, Hud NV (2015) Ester-mediated amide bond formation driven by wet–dry cycles: a possible path to polypeptides on the prebiotic earth. Angew Chem Int Ed 54:9871–9875
Fox S, Strasdeit H (2013) Possible prebiotic origin on volcanic islands of oligopyrrole-type photopigments and electron transfer cofactors. Astrobiology 13:578–595
Fox S, Strasdeit H (2017) Inhabited or uninhabited? Pitfalls in the interpretation of possible chemical signatures of extraterrestrial life. Front Microbiol 8:1622
Fox S, Pleyer HL, Strasdeit H (2018) An automated apparatus for the simulation of prebiotic wet–dry cycles under strictly anaerobic conditions. Int J Astrobiol 1–13, available under First View at https://doi.org/10.1017/S1473550418000010
Fraústo da Silva JJR, Williams RJP (2001) The biological chemistry of the elements, 2nd edn. Oxford University Press, Oxford, pp 436–449
Frydman RB, Stevens E (1968) Non-enzymatic chelation of divalent metals with uroporphyrins under physiological conditions. Biochim Biophys Acta 165:167–169
Gill R (2010) Igneous rocks and processes: a practical guide. Wiley-Blackwell, Chichester Fig. 5.5.1 on p 149
Glemser O, Schwarzmann E (1981) Kobalt, Nickel. In: Brauer G (ed) Handbuch der Präparativen Anorganischen Chemie, vol 3, 3rd edn. Ferdinand Enke, Stuttgart, pp 1659–1703
Goff HM (1981) Iron(III) porphyrin–imidazole complexes. Analysis of carbon-13 nuclear magnetic resonance isotropic shifts and unpaired spin delocalization. J Am Chem Soc 103:3714–3722
Hazen RM (2013) Paleomineralogy of the Hadean eon: a preliminary species list. Am J Sci 313:807–843
Hodgson GW, Baker BL (1967) Porphyrin abiogenesis from pyrrole and formaldehyde under simulated geochemical conditions. Nature 216:29–32
Hofmann BA, Lorenzetti S, Eugster O, Krähenbühl U, Herzog G, Serefiddin F, Gnos E, Eggimann M, Wasson JT (2009) The Twannberg (Switzerland) IIG iron meteorites: mineralogy, chemistry, and CRE ages. Meteorit Planet Sci 44:187–199
Holland HD (1973) The oceans: a possible source of iron in iron-formations. Econ Geol 68:1169–1172
Holland HD (1984) The chemical evolution of the atmosphere and oceans. Princeton University Press, Princeton, p 110
Ivashin NV, Shulga AM, Terekhov SN, Dzilinski K (1996) Physical and chemical transformations of μ-oxo dimers and alkoxy complexes of Fe-octaethylporphyrins in solids and in solutions. Spectrochim Acta A 52:1603–1614
Izawa MRM, Nesbitt HW, MacRae ND, Hoffman EL (2010) Composition and evolution of the early oceans: evidence from the Tagish Lake meteorite. Earth Planet Sci Lett 298:443–449
James BR (1978) Interaction of dioxygen with metalloporphyrins. In: Dolphin D (ed) The porphyrins, vol 5. Academic Press, New York, pp 205–302
Johnson EC, Dolphin D, Cushing MA Jr, Ittel SD (1980) Metalloporphines. In: Busch DH (ed) Inorganic syntheses, vol 20. Wiley, New York, pp 143–147
Kaim W, Schwederski B, Klein A (2013) Bioinorganic chemistry: inorganic elements in the chemistry of life, 2nd edn. Wiley, Chichester, pp 22–31, 37–116, 172–177
Kalish HR, Latos-Grażyński L, Balch AL (2000) Heme/hydrogen peroxide reactivity: formation of paramagnetic iron oxophlorin isomers by treatment of iron porphyrins with hydrogen peroxide. J Am Chem Soc 122:12478–12486
Kalish H, Camp JE, Stȩpień M, Latos-Grażyński L, Balch AL (2001) Reactivity of mono-meso-substituted iron(II) octaethylporphyrin complexes with hydrogen peroxide in the absence of dioxygen. Evidence for nucleophilic attack on the heme. J Am Chem Soc 123:11719–11727
Kareem K (2005) Komatiites of the Weltevreden formation, Barberton Greenstone Belt, South Africa: implications for the chemistry and temperature of the Archean mantle. Dissertation. Louisiana State University, 227 pp
Khosropour R, Hambright P (1972) A general mechanism for metal ion incorporation into porphyrin molecules. J Chem Soc Chem Commun:13–14
Knauth LP (1998) Salinity history of the Earth’s early ocean. Nature 395:554–555
Knauth LP (2005) Temperature and salinity history of the Precambrian Ocean: implications for the course of microbial evolution. Palaeogeogr Palaeoclimatol Palaeoecol 219:53–69
Konarev DV, Khasanov SS, Saito G, Lyubovskaya RN (2009) Design of molecular and ionic complexes of fullerene C60 with metal(II) octaethylporphyrins, MIIOEP (M = Zn, Co, Fe, and Mn) containing coordination M–N(ligand) and M–C(C60 −) bonds. Cryst Growth Des 9:1170–1181
Kumar P, Nagarajan R, Sarangi R (2013) Quantitative X-ray absorption and emission spectroscopies: electronic structure elucidation of Cu2S and CuS. J Mater Chem C 1:2448–2454
Lahav N, White DH (1980) A possible role of fluctuating clay-water systems in the production of ordered prebiotic oligomers. J Mol Evol 16:11–21
Lahav N, White D, Chang S (1978) Peptide formation in the prebiotic era: thermal condensation of glycine in fluctuating clay environments. Science 201:67–69
Lathe R (2004) Fast tidal cycling and the origin of life. Icarus 168:18–22
Lathe R (2006) Early tides: response to Varga et al. Icarus 180:277–280
Li W, Czaja AD, Van Kranendonk MJ, Beard BL, Roden EE, Johnson CM (2013) An anoxic, Fe(II)-rich, U-poor ocean 3.46 billion years ago. Geochim Cosmochim Acta 120:65–79
Liang W, Whangbo M-H (1993) Conductivity anisotropy and structural phase transition in covellite CuS. Solid State Commun 85:405–408
Lindsey JS, Ptaszek M, Taniguchi M (2009) Simple formation of an abiotic porphyrinogen in aqueous solution. Orig Life Evol Biosph 39:495–515
Lindsey JS, Chandrashaker V, Taniguchi M, Ptaszek M (2011) Abiotic formation of uroporphyrinogen and coproporphyrinogen from acyclic reactants. New J Chem 35:65–75
Mamajanov I, MacDonald PJ, Ying J, Duncanson DM, Dowdy GR, Walker CA, Engelhart AE, Fernández FM, Grover MA, Hud NV, Schork FJ (2014) Ester formation and hydrolysis during wet−dry cycles: generation of far-from-equilibrium polymers in a model prebiotic reaction. Macromolecules 47:1334–1343
Maricondi C, Swift W, Straub DK (1969) Thermomagnetic analysis of hemin and related compounds. J Am Chem Soc 91:5205–5210
Mazin II (2012) Structural and electronic properties of the two-dimensional superconductor CuS with
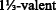 copper. Phys Rev B 85:115133
copper. Phys Rev B 85:115133Nečas D, Klapetek P (2012) Gwyddion: an open-source software for SPM data analysis. Cent Eur J Phys 10:181–188
Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW, Feyereisen R, Gonzalez FJ, Coon MJ, Gunsalus IC, Gotoh O, Okuda K, Nebert DW (1993) The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names of enzymes, and nomenclature. DNA Cell Biol 12:1–51
Nozaki H, Shibata K, Ohhashi N (1991) Metallic hole conduction in CuS. J Solid State Chem 91:306–311
Ogoshi H, Watanabe E, Yoshida Z (1973) Porphyrin acids. Tetrahedron 29:3241–3245
Olasagasti F, Kim HJ, Pourmand N, Deamer DW (2011) Non-enzymatic transfer of sequence information under plausible prebiotic conditions. Biochimie 93:556–561
Pohorille A (2009) Early ancestors of existing cells. In: Rasmussen S, Bedau MA, Chen L, Deamer D, Krakauer DC, Packard NH, Stadler PF (eds) Protocells: bridging nonliving and living matter. MIT Press, Cambridge, pp 563–581
Puchtel IS, Blichert-Toft J, Touboul M, Walker RJ, Byerly GR, Nisbet EG, Anhaeusser CR (2013) Insights into early earth from Barberton komatiites: evidence from lithophile isotope and trace element systematics. Geochim Cosmochim Acta 108:63–90
Ralphs K, Zhang C, James SL (2017) Solventless mechanochemical metallation of porphyrins. Green Chem 19:102–105
Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R (2004) Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem 331:370–375
Rodriguez-Garcia M, Surman AJ, Cooper GJT, Suárez-Marina I, Hosni Z, Lee MP, Cronin L (2015) Formation of oligopeptides in high yield under simple programmable conditions. Nat Commun 6:8385
Saetia S, Liedl KR, Eder AH, Rode BM (1993) Evaporation cycle experiments – a simulation of salt-induced peptide synthesis under possible prebiotic conditions. Orig Life Evol Biosph 23:167–176
Saito MA, Sigman DM, Morel FMM (2003) The bioinorganic chemistry of the ancient ocean: the co-evolution of cyanobacterial metal requirements and biogeochemical cycles at the Archean–Proterozoic boundary? Inorg Chim Acta 356:308–318
Schneider J, Franke M, Gurrath M, Röckert M, Berger T, Bernardi J, Meyer B, Steinrück H-P, Lytken O, Diwald O (2016) Porphyrin metalation at MgO surfaces: a spectroscopic and quantum mechanical study on complementary model systems. Chem Eur J 22:1744–1749
Soares ARM, Taniguchi M, Chandrashaker V, Lindsey JS (2012a) Primordial oil slick and the formation of hydrophobic tetrapyrrole macrocycles. Astrobiology 12:1055–1068
Soares ARM, Taniguchi M, Chandrashaker V, Lindsey JS (2012b) Self-organization of tetrapyrrole constituents to give a photoactive protocell. Chem Sci 3:1963–1974
Soares ARM, Taniguchi M, Chandrashaker V, Lindsey JS (2013a) Expanded combinatorial formation of porphyrin macrocycles in aqueous solution containing vesicles. A prebiotic model. New J Chem 37:1073–1086
Soares ARM, Anderson DR, Chandrashaker V, Lindsey JS (2013b) Catalytic diversification upon metal scavenging in a prebiotic model for formation of tetrapyrrole macrocycles. New J Chem 37:2716–2732
Staudacher T, Ferrazzini V, Peltier A, Kowalski P, Boissier P, Catherine P, Lauret F, Massin F (2009) The April 2007 eruption and the Dolomieu crater collapse, two major events at Piton de la Fournaise (La Réunion Island, Indian Ocean). J Volcanol Geotherm Res 184:126–137
Strasdeit H, Fox S (2013) Experimental simulations of possible origins of life: conceptual and practical issues. In: de Vera J-P, Seckbach J (eds) Habitability of other planets and satellites. Springer, Dordrecht, pp 129–144
Szutka A (1964) Porphine-like substances: probable synthesis during chemical evolution. Nature 202:1231–1232
Taniguchi M, Soares ARM, Chandrashaker V, Lindsey JS (2012) A tandem combinatorial model for the prebiogenesis of diverse tetrapyrrole macrocycles. New J Chem 36:1057–1069
Varga P, Rybicki KR, Denis C (2006) Comment on the paper “fast tidal cycling and the origin of life” by Richard Lathe. Icarus 180:274–276
Walker JCG (1982) Climatic factors on the Archean earth. Palaeogeogr Palaeoclimatol Palaeoecol 40:1–11
Walker JCG, Klein C, Schidlowski M, Schopf JW, Stevenson DJ, Walter MR (1983) Environmental evolution of the Archean–early Proterozoic earth. In: Schopf JW (ed) Earth’s earliest biosphere: its origin and evolution. Princeton University Press, Princeton, pp 260–263
Wells AF (1984) Structural inorganic chemistry, 5th edn. Clarendon Press, Oxford, pp 1142–1144
Wiberg N (ed) (2001) Holleman-Wiberg inorganic chemistry. Academic Press, San Diego, pp 195–1057
Wood TE, Thompson A (2007) Advances in the chemistry of dipyrrins and their complexes. Chem Rev 107:1831–1861
Zahnle K, Walker JCG (1987) A constant daylength during the Precambrian era? Precambrian Res 37:95–105
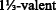 copper. Phys Rev B 85:115133
copper. Phys Rev B 85:115133Acknowledgements
The authors are grateful to Thomas Staudacher (Observatoire volcanologique du Piton de la Fournaise, La Réunion) for assistance with sampling of the basalt and to Dr. Manfred Martin (Regierungspräsidium Freiburg; Landesamt für Geologie, Rohstoffe und Bergbau) for the basalt analysis. We also thank Dr. Beda Hofmann (Naturhistorisches Museum Bern) for the meteorite samples and Dr. Igor Puchtel (University of Maryland, College Park) for the komatiite. Special thanks are due to Prof. Wolfgang Hanke and Prof. Wolfgang Schwack (both at the Universität Hohenheim, Stuttgart) who provided the fluorescence microscopy and HPTLC equipment, respectively, and helped us with the measurements. We are also grateful to Dr. Frank Trixler (Deutsches Museum, München; School of Education, Technische Universität München; Center for NanoScience (CeNS), Ludwig-Maximilians-Universität München) who provided HLP the opportunity to perform AFM measurements during a research stay in Munich. We thank Sonja Ringer for technical assistance. HLP thanks the State of Baden-Württemberg for an LGFG doctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pleyer, H.L., Strasdeit, H. & Fox, S. A Possible Prebiotic Ancestry of Porphyrin-Type Protein Cofactors. Orig Life Evol Biosph 48, 347–371 (2018). https://doi.org/10.1007/s11084-018-9567-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-018-9567-4