
Key message
Application of a low-input chromatin profiling method, CUT&RUN, to FACS-purified Arabidopsis endosperm nuclei generates parental-specific genome-wide H3K27me3 landscapes with high sensitivity, specificity and reproducibility.
Abstract
Endosperm is an essential seed tissue with a unique epigenetic landscape. During endosperm development, differential epigenetic regulation of the maternal and paternal genomes plays important roles in regulating gene expression, especially at imprinted genes. In Arabidopsis, profiling the epigenetic landscape of endosperm on a genome-wide scale is challenging due to its small size, mode of development and close association with maternal tissue. Here, we applied a low-input chromatin profiling method, CUT&RUN (cleavage under targets and release using nuclease), to profile parental-specific chromatin modifications using limited numbers of Arabidopsis endosperm nuclei. We demonstrate that CUT&RUN generates genome-wide H3K27me3 landscapes with high sensitivity, specificity and reproducibility using around 20,000 endosperm nuclei purified by flow cytometry and fluorescence-activated cell sorting. H3K27me3 peaks identified by CUT&RUN and previous ChIP (chromatin immunoprecipitation) approaches were largely overlapping, with some distinctions in heterochromatin. The versatility and simplicity of CUT&RUN make it a viable alternative to ChIP, which requires greater amounts of starting material, and will enable further study of tissue- or cell-type-specific epigenomes in Arabidopsis and other plant species.
Similar content being viewed by others


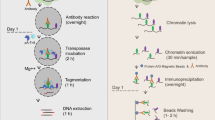
Introduction
Endosperm is an essential seed tissue formed during flowering plant reproduction. During double fertilization, the two sperms from one pollen grain fertilize the female gametes, the egg cell and the central cell, forming embryo and endosperm, respectively. The endosperm functions to support embryo growth and development. Epigenetic regulators play essential roles in endosperm and seed development. Central cell-specific activity of the DNA demethylase DEMETER establishes widespread differences in DNA methylation patterns between the maternal and paternal genomes before fertilization (Gehring et al. 2006, 2009; Hsieh et al. 2009; Park et al. 2016). DNA hypomethylation of maternally inherited genomes is required for proper endosperm gene expression programs, especially at imprinted genes (Gehring et al. 2009; Hsieh et al. 2011). Imprinting refers to gene expression derived predominantly from one of the parental alleles (Gehring 2013). In addition to differences in DNA methylation, differences in histone modifications between maternally and paternally inherited alleles are also important for maintaining imprinted gene expression (Gehring 2013). The parental-specific expression pattern of some imprinted genes is disrupted in a PRC2 mutant (Hsieh et al. 2011) and PRC2 mutants exhibit abnormal endosperm development that leads to seed abortion (Grossniklaus et al. 1998; Luo et al. 1999; Ohad et al. 1996). PRC2 is required for trimethylation at lysine 27 of histone 3 (H3K27me3). Examination of endosperm DNA methylation and histone states has revealed a strong correlation between paternally expressed imprinted genes (PEGs), parental differences in DNA methylation patterns and the distribution of H3K27me3 (Moreno-Romero et al. 2016; Pignatta et al. 2014). For example, PHE1 is an imprinted gene that is preferentially expressed from the paternal allele. The PHE1 flanking DNA is more methylated on the paternal allele, whereas H3K27me3 is enriched on the silent maternal allele (Hsieh et al. 2011; Makarevich et al. 2008; Moreno-Romero et al. 2016; Wolff et al. 2011). These observations, along with those in other species, have demonstrated the dynamic nature of epigenetic marks in endosperm. Therefore, parental-specific profiles of epigenetic marks at different developmental stages could help illuminate the molecular mechanisms underlying gene imprinting and endosperm gene expression programs more generally.
Yet, it has been technically challenging to profile histone modifications in endosperm, particularly in species like Arabidopsis thaliana with small, ephemeral endosperm. Currently, the most common method for chromatin modification profiling is chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq), which requires large amounts of starting material. Each developing Arabidopsis seed only contains hundreds of endosperm nuclei (Boisnard-Lorig et al. 2001), making it a challenging task to isolate enough endosperm nuclei for ChIP-seq. By applying INTACT (isolation of nuclei tagged in specific cell types; Deal and Henikoff 2010) to purify endosperm nuclei, ChIP-seq was previously successfully performed to examine epigenetic marks (H3K27me3, H3K27me1 and H3K9me2) in endosperm using around 500,000 endosperm nuclei (Moreno-Romero et al. 2016, 2017). However, the adoption of INTACT for ChIP-seq has some limitations. To obtain enough seeds at a specific developmental stage, male-sterile mutants were used to facilitate crosses (Moreno-Romero et al. 2016). Transformation with BirA and NTF constructs is required by the principles of INTACT (Deal and Henikoff 2010), and specificity of the nuclei purified by INTACT is limited by the expression pattern of the promoter used to drive the expression of BirA and NTF. Therefore, construction of transgenic lines harboring all of the required mutations and transgenes, along with the restriction imposed by promoter choice, hinders the application of INTACT-ChIP-seq to readily profile the epigenome in different developmental stages and in different mutant backgrounds.
Desiring a versatile method for profiling histone states in endosperm, we applied the newly developed CUT&RUN (cleavage under targets and release using nuclease) method to Arabidopsis (Skene et al. 2018; Skene and Henikoff 2017). CUT&RUN utilizes a different strategy from ChIP to profile genome-wide DNA binding sites of a protein of interest. CUT&RUN targets micrococcal nuclease (MNase) to binding sites of the protein of interest through specific interactions between the protein of interest, a primary antibody and protein A fused with MNase. When MNase activity is activated by Ca2+ addition, MNase cleaves DNA around the binding sites, generating small DNA fragment–protein complexes. The reaction is kept at 0 °C to minimize diffusion and secondary cutting by MNase, and stopped at the designated time by chelation. The small DNA–protein complexes diffuse through nuclear pore and are released to the solution. After spinning down the nuclei, the small DNA fragments in the solution are then purified and prepared for sequencing (Table 1). The majority of the unbound DNA remains in the nuclei and is left out from sequencing library preparation. Thus, CUT&RUN sequencing data have high signal-to-background noise ratio and the procedure requires lower sequencing depth compared to ChIP. In addition, utilization of concanavalin A-coated magnetic beads allows efficient capture and retention of nuclei because of the interaction between concanavalin A and glycoproteins in the nuclear membrane. CUT&RUN has been shown to successfully profile DNA binding sites of transcription factors from 1000 mammalian cells and histone modification landscapes from just 100 mammalian cells (Skene et al. 2018). Taken together, the amount of nuclei needed to profile the DNA binding sites of a protein of interest is significantly reduced compared to ChIP (Skene and Henikoff 2017). In addition, CUT&RUN does not require construction of specific transgenic lines, except that when a primary antibody against the protein of interest is not available, transgenic lines harboring an epitope-tagged protein construct are necessary (ex. FLAG-tagged isoforms). Therefore, if successfully applied to Arabidopsis, CUT&RUN could facilitate epigenome profiling of various developmental stages and in various mutant backgrounds. Moreover, the same method could also be readily applied to other plant species for epigenome profiling.
In this study, we demonstrate the application of CUT&RUN to epigenome profiling using nuclei purified by flow cytometry and fluorescence-activated cell sorting (FACS). When applied to 20,000 leaf nuclei, CUT&RUN generated a H3K27me3 profile largely overlapping the profile obtained from leaf ChIP. When applied to 20,000 endosperm nuclei, CUT&RUN revealed about 65% of the H3K27me3 endosperm peaks previously identified by INTACT-ChIP on 500,000 endosperm nuclei. Distinct H3K27me3 peaks that were not detected by INTACT-ChIP were also identified. By combining sequencing data from four independent replicates (from a total of 80,000 nuclei), CUT&RUN revealed parental-specific biases in H3K27me3 distribution.
Results
Ploidy-based FACS isolates endosperm nuclei from embryo and seed coat nuclei
To isolate endosperm nuclei from seeds, we sorted seed nuclei based on DNA content using FACS. Triploidy is a signature of Arabidopsis endosperm, which has two identical maternal genomes and one paternal genome. The other two seed tissues, embryo and seed coat, are both diploid, allowing endosperm nuclei to be distinguished from the rest of seed nuclei. Nuclei with differing DNA content can be separated by their DAPI intensity (Fig. 1a). 2C, 4C and 8C nuclei represent the diploid embryo and seed coat nuclei, while 3C and 6C nuclei represent the triploid endosperm nuclei. The dominance of the higher-order ploidy peaks (4C and 6C) presumably represents active DNA replication and nuclear division in young seeds (at 4 days after pollination, the embryo was at the globular stage under our growth conditions). The 3C peak, which corresponds to un-replicated endosperm nuclei, was difficult to cleanly separate from the 4C peak at this stage. Therefore, only 6C nuclei representing replicated endosperm nuclei were collected for endosperm epigenome profiling.

CUT&RUN reproduces the leaf H3K27me3 landscape with high accuracy and sensitivity using a small number of nuclei. a Histogram shows FACS gated nuclei with different DAPI intensity. The nuclei were extracted from whole seeds from crosses between Col and Ler at 4 days after pollination. The plausible ploidy of each peak is indicated. b Snapshot of results from CUT&RUN performed on 200,000 or 50,000 FACS-sorted embryo and seed coat nuclei (2C, 4C and 8C from DAPI-based FACS). MNase digestion time varied from 15 min to 2 min. For comparison, published leaf H3K27me3 ChIP data (GSE66585) were reanalyzed in the same manner. c Normalized reads from CUT&RUN with H3K27me3 antibody or no antibody experiments were aligned over H3K27me3 or H3K27me1 ChIP peaks. Only H3K27me1 ChIP peaks that did not overlap H3K27me3 peaks were included. Regions 2000 bp upstream and downstream from the H3K27me3 peaks and H3K27me1 peaks are also shown. RPGC, reads per genomic content. d Normalized reads from CUT&RUN with H3, H3K27me3 and IgG antibodies on leaf nuclei were aligned over leaf H3K27me3 ChIP peaks. The average signals inside and 2 kb upstream and downstream of the peaks were plotted above the heatmaps. The reads were normalized to have 1x genomic content for each library. Therefore, IgG data, which had more uncovered 150-bp bins, display much higher average signals than H3, which had high read coverage over the entire genome. The peaks in all three heatmaps are in the same order and ranked by decreasing CUT&RUN H3K27me3 occupancy. RPGC, reads per genomic content. e Ratio between H3K27me3 and H3 normalized reads in 150-bp bins were plotted over leaf H3K27me3 ChIP peaks. Missing data in all heatmaps were colored black. S start of peak region; E end of peak region. f Venn diagram shows the overlap between leaf H3K27me3 peaks called from CUT&RUN and ChIP
CUT&RUN reproduces the leaf H3K27me3 landscape with high accuracy and sensitivity
We first applied CUT&RUN to profile the H3K27me3 landscape using embryo and seed coat nuclei from FACS (2C, 4C and 8C nuclei) to compare to published ChIP data from leaf tissue (GSE66585; Moreno-Romero et al. 2016). We tested CUT&RUN with 200,000 and 50,000 nuclei. To optimize experimental conditions, we also tested two different MNase active digestion times (15 min and 2 min). As shown in Fig. 1b, CUT&RUN H3K27me3 profiles were similar to ChIP, exhibiting specific peaks at the ChIP H3K27me3 peak regions. To validate whether CUT&RUN is sensitive to detect as many peaks as ChIP, we analyzed the normalized reads obtained by CUT&RUN over all H3K27me3 peaks identified in leaf ChIP (Fig. 1c). Almost all H3K27me3 ChIP peaks exhibited increased H3K27me3 CUT&RUN signals relative to the flanking regions (Fig. 1c). For regions enriched for H3K27me1 in ChIP, no increased signals were observed in the H3K27me3 CUT&RUN profile, as expected (Fig. 1c). As a control, we also performed CUT&RUN without a primary antibody. There was no enrichment over the two leaf histone modification peaks in these data (Fig. 1c). Together, these results demonstrate the specificity and sensitivity of CUT&RUN. Starting with as few as 50,000 nuclei achieved the same sensitivity as 200,000 nuclei, even with a shorter MNase active digestion time of 2 min (Fig. 1c).
After testing additional conditions, we found that as few as 5000 nuclei with a MNase active digestion time of 30 min generated similar H3K27me3 profiles as 50,000 nuclei (data not shown). Starting with the same number of nuclei, CUT&RUN with a histone 3 (H3) antibody reproducibly generated concentrated libraries with high complexity, while IgG negative control CUT&RUN generated low-concentration libraries with low complexity (Table S1). We thus decided to use about 3000 nuclei to profile H3 and about 20,000 nuclei to profile modified histones and for the IgG negative control (Table S1). To further validate the CUT&RUN method in plants, we performed CUT&RUN on leaf nuclei to directly compare to the leaf ChIP-seq dataset (GSE66585). We used the same program, epic (“epic: diffuse domain ChIP-Seq caller based on SICER,” https://github.com/endrebak/epic; Zang et al. 2009), to call peaks from the CUT&RUN data and from the leaf H3K27me3 ChIP-seq dataset (GSE66585). CUT&RUN performed with the H3K27me3 antibody generated specific signals over almost all leaf ChIP H3K27me3 peaks, while no enriched signals were observed in these same regions when H3 and IgG antibodies were used (Fig. 1d). The H3K27me3/H3 ratio was similarly enriched in ChIP H3K27me3 peaks, while the flanking regions were not (Fig. 1e). 94% of the H3K27me3 peaks identified using CUT&RUN were also identified from H3K27me3 ChIP (Fig. 1f and Table S2). These results demonstrate that CUT&RUN is a viable alternative to ChIP.
CUT&RUN using endosperm nuclei revealed both similar and distinct H3K27me3 peaks compared to INTACT-ChIP
Having demonstrated that CUT&RUN works using FACS-sorted Arabidopsis nuclei, we performed CUT&RUN on endosperm nuclei to generate H3K27me3 landscapes. We collected 6C nuclei from seeds of crosses between Col and Ler at the same time after pollination (4 days) as that used in the only published endosperm H2K37me3 INTACT-ChIP experiments (Moreno-Romero et al. 2016, 2017). When the 6C nuclei collected from FACS were reanalyzed using flow cytometry, 17% of the collected nuclei were revealed to be either 2C or 4C nuclei (Table S3), demonstrating that, under our experimental conditions, contamination from seed coat and embryo was around 17% for endosperm nuclei at 4 DAP. We analyzed CUT&RUN and INTACT-ChIP data (GSE66585) in the same manner and called H3K27me3 peaks by comparing the ratio of H3K27me3 to H3 with the program epic (https://github.com/endrebak/epic; Zang et al. 2009). H3K27me3 CUT&RUN profiles exhibited higher signals over the INTACT-ChIP H3K27me3 peaks relative to the flanking regions, while H3 and IgG signals were not enriched in the peak regions (Fig. 2a). Endosperm CUT&RUN exhibited increased H3K27me3 signals in about two thirds of endosperm INTACT-ChIP peaks (Fig. 2a). Enrichment of H3K27me3 relative to H3 revealed the same result (Fig. 2b). CUT&RUN data generated similar number of H3K27me3 peaks as INTACT-ChIP, and the overlap between CUT&RUN peaks and INTACT-ChIP peaks was around 65% (Fig. 2c and Table S2). Although CUT&RUN did not detect about 35% of INTACT-ChIP peak regions (Fig. 2d), in many cases the CUT&RUN H3K27me3 profiles appear to be quite similar to the INTACT-ChIP profiles, but with a lower read coverage (Fig. 2d right panel). These 35% of INTACT-ChIP peaks were not peaks that showed relatively low H3K27me3 occupancy in INTACT-ChIP (Figure S1). Meanwhile, around 30% of CUT&RUN peaks were not identified by INTACT-ChIP, where CUT&RUN showed higher sensitivities (Fig. 2d, left panel). When compared to H3K27me3 ChIP data from other tissues, 39% of the genes uniquely identified as marked by H3K27me3 in endosperm CUT&RUN were detected as marked by H3K27me3 from H3K27me3 ChIP on Arabidopsis seedlings (Zhang et al. 2007) and 44% of them overlapped with H3K27me3 peaks identified in ChIP-seq data on germinating Arabidopsis embryos (Xiao et al. 2017). The overlap with these two ChIP datasets was significant based on hypergeometric tests (p value: 1.5e-127 and 7.5e-176, respectively).

Endosperm CUT&RUN reveals similar and distinct H3K27me3 peaks compared to INTACT-ChIP. a Normalized reads with H3, H3K27me3 and IgG antibodies from CUT&RUN on unfixed endosperm nuclei (Endo_Unfixed_H3_1, Endo_Unfixed_H3K27me3_1, Endo_Unfixed_IgG_1 in Table S1) were aligned over endosperm H3K27me3 INTACT-ChIP peaks. The average signals inside and 2 kb upstream and downstream of the peaks were plotted above the heatmaps. The reads were normalized to have 1x genomic content for each library. Therefore, IgG data, which had more uncovered 150-bp bins, display much higher average signals than H3, which had high read coverage over the entire genome. The peaks in all three heatmaps are in the same order and ranked by decreasing CUT&RUN H3K27me3 occupancy. Endosperm H3K27me3 INTACT-ChIP data (GSE66585) were reanalyzed in the same manner as CUT&RUN data. RPGC, reads per genomic content. b Ratio between H3K27me3 and H3 normalized reads in 150-bp bins were plotted over endosperm H3K27me3 INTACT-ChIP peaks. The average ratio inside and 2 kb upstream and downstream of the peaks were plotted above the heatmaps. The peaks in the heatmap were ordered by CUT&RUN H3K27me3 occupancy in each experiment. Missing data in the heatmaps were colored black. Unfixed, data from CUT&RUN using nuclei without formaldehyde fixation (Endo_Unfixed_1 in Table S1). Fixed, CUT&RUN data using formaldehyde-fixed nuclei (Endo_Fixed_1 in Table S1). Merged, pooled data from four independent CUT&RUN replicates. S start of peak region; E end of peak region. c Venn diagram shows the overlaps between H3K27me3 peaks called from different experiments. d Browser tracks comparing H3K27me3 normalized reads from INTACT-ChIP, different CUT&RUN experiments using unfixed or fixed nuclei, and a merged dataset from four independent CUT&RUN experiments. Orange and purple bars represent H3K27me3 peaks called from INTACT-ChIP and CUT&RUN, respectively
Although CUT&RUN does not require formaldehyde fixation, we tested the effect of formaldehyde fixation on CUT&RUN profiles. It was possible that the small DNA fragment–protein complexes generated by protein A–MNase digestion might not be able to easily diffuse out of fixed nuclei (Skene and Henikoff 2017). Thus, for the fixed preps we extracted total DNA from digested nuclei and then size-selected small DNA fragments (less than 1000 bp). Overall, fixed nuclei gave very similar results to unfixed nuclei (Fig. 2b–d and S2). H3K27me3 peaks called from fixed and unfixed CUT&RUN were largely overlapping (over 88%; Fig. 2c and Table S2). CUT&RUN data on fixed nuclei exhibited slightly broader peaks than that from unfixed nuclei, which could be explained by more rigid DNA–protein conformations in fixed nuclei limiting the digestion of unprotected DNA. Alternatively, the total DNA extraction method could introduce more background (Fig. 2b and S2A).
Comparison between independent CUT&RUN experiments indicated the reproducibility of CUT&RUN: two independent experiments on unfixed nuclei and two on fixed nuclei showed similar enrichment profiles in all peaks when the peaks were displayed in the same order (Figure S2A). Pearson correlation analysis on genome-wide read coverage in 10-kb windows for each library confirmed the similarity of the profiles (Figure S2B). The merged data from all four replicates exhibited the same profiles as each individual replicate, demonstrating that the sequencing depth of each individual experiment (with around 5 million reads from about 20,000 nuclei) is sufficient for identifying overall H3K27me3 peaks (Fig. 2b, d and Table S2).
CUT&RUN was reported to reveal transcription factor binding sites with significantly higher resolution than ChIP (Skene and Henikoff 2017). We examined the spread of H3K27me3 signals from the centers of peaks (overlapping peaks from CUT&RUN and ChIP) using the same number of unique deduplicated reads from either CUT&RUN or ChIP (Figure S3). The CUT&RUN H3K27me3 signal exhibited a slightly narrower spread over peaks compared to that from ChIP in both leaf and endosperm samples (Figure S3A, B and S3D, E). The distributions of peak length were also similar between CUT&RUN and ChIP, with a mean length of around 4 kb in leaf samples and around 2.5 kb in endosperm samples (Figure S3C and F). These comparisons suggest that CUT&RUN and ChIP performed similarly in revealing the boundaries of broad histone modification peaks such as H3K27me3.
H3K27me3 profiles from CUT&RUN and INTACT-ChIP on endosperm nuclei differed in the pericentromeric regions
To further examine the differences observed between CUT&RUN and INTACT-ChIP data, we analyzed the chromosomal distribution of the H3K27me3/H3 ratio and plotted the Z-score (Fig. 3a). Whereas the two CUT&RUN experiments overlapped with INTACT-ChIP data in the chromosomal arms, the number of CUT&RUN and INTACT-ChIP peaks differed substantially in the pericentromeric regions. Endosperm INTACT-ChIP identified many H3K27me3 peaks in pericentromeric regions, which were not observed in leaf ChIP data (Moreno-Romero et al. 2016). CUT&RUN detected only a small portion of these endosperm pericentromeric peaks (Fig. 2d right panel, Fig. 3). The chromosomal distribution of both H3K27me3 and H3 differed from the ones obtained by INTACT-ChIP in the pericentromeric regions (Fig. 3b, c). CUT&RUN H3K27me3 exhibited a significant decrease in coverage around pericentromeric regions, whereas H3 signals were fairly constant across the chromosome (Fig. 3b, c). By contrast, pericentromeric H3K27me3 INTACT-ChIP signals were not depleted in the pericentromeric regions but H3 signals were (Fig. 3b, c). Thus, CUT&RUN H3K27me3 and H3 profiles differ from their respective ChIP profiles in opposite ways, exacerbating difference in the Z-score of log2 H3K27me3/H3 ratio and the peaks detected around pericentromeric regions (Fig. 3a–c). By contrast, both H3K27me3 and H3 profiles from CUT&RUN were highly similar to that from ChIP in leaf samples (Fig. 3e). CUT&RUN using the negative control IgG antibody, which reveals the cutting efficiency of MNase, generated relatively uniform coverage across the whole chromosome (Fig. 3d). These results suggest that the decreased H3K27me3 signals in pericentromeric regions were not caused by reduced accessibility of heterochromatic regions to MNase. Despite the difference in peak calling, visual examination of the INTACT-ChIP and CUT&RUN data from endosperm in these regions indicated similar profiles but with lower read coverage/signal from CUT&RUN (Fig. 2d right panel).

CUT&RUN and INTACT-ChIP on endosperm nuclei differed in the pericentromeric regions. Chromosomal view of a Z-score of the log2-transformed ratio between H3K27me3 and H3, b–d normalized reads of b H3K27me3, c H3 and d IgG from two CUT&RUN experiments and INTACT-ChIP (GSE66585) are shown across chromosome 1. Note that the two CUT&RUN experiments were mostly overlapping so that one of them is hardly visible. e Chromosomal view of normalized reads of H3K27me3 and H3 from CUT&RUN and ChIP (GSE66585) from leaf samples. Brown box marks the position of centromere. RPGC, reads per genomic content
Parental biases of H3K27me3 could be identified using CUT&RUN data
To analyze whether the H3K27me3 modification was biased toward the maternally or paternally inherited genomes in specific regions, as has been suggested by INTACT-ChIP results, we merged all four CUT&RUN experiments and identified allele-specific reads from the pooled data (Table S1). In this experiment, Col reads are derived from the maternally inherited genome and Ler reads are from the paternally inherited genome. The Col fraction of allele-specific reads was calculated for H3 and H3K27me3 in 150-bp windows. The mean Col fraction of H3 reads was 0.8 instead of the expected 0.67, suggesting some mapping bias to Col and contamination from non-endosperm nuclei (Fig. 4a, Table S1 and Table S3). However, the Col fraction of H3K27me3 was significantly skewed to the right compared to H3 (Fig. 4a and Table S1), revealing that the deposition of H3K27me3 is heavily biased to the maternally inherited Col genome. To identify specific regions with Col-biased H3K27me3, we first filtered out the top and bottom 10% of H3 bins exhibiting allelic bias, to retain only those bins that did not exhibit significant H3 allelic bias (Fig. 4a). From the remaining bins, we classified those bins with an H3K27me3 Col fraction larger than the 90th percentile of the H3 distribution as Col-biased, whereas bins with H3K27me3 Col fraction smaller than the 10th percentile of the H3 distribution were classified as Ler-biased (Fig. 4a). While Col-biased regions were widely distributed across chromosomal arms, Ler-biased regions accumulated in the pericentromeric regions (Fig. 4b). We further identified Col- or Ler-biased H3K27me3 peaks by intersecting parentally biased regions and H3K27me3 peaks identified in Fig. 2 (Fig. 4c).

Parental biases of H3K27me3 identified using CUT&RUN data. a The top histogram shows the H3 Col fraction for 150-bp bins that had at least 20 total allele-specific reads. The 10th and 90th percentile cutoffs are shown. Only the bins between the cutoffs were kept for the H3K27me3 Col fraction analysis, shown in the bottom histogram. b Bar graph showing the distribution of the Col- and Ler-biased H3K27me3 regions in each 10-kb window across chromosome 5. c H3K27me3 peaks that overlapped with Col- or Ler-biased H3K27me3 regions were considered parentally biased H3K27me3 peaks. Bar graph revealed the frequency of the Col- and Ler-biased H3K27me3 peaks in each 10-kb window across chromosome 5
Prior H3K27me3 INTACT-ChIP data from endosperm demonstrated that most PEGs were associated with maternal H3K27me3 peaks but not paternal H3K27me3 peaks. In contrast, maternally expressed imprinted genes (MEGs) were rarely marked with paternal H3K27me3 (Moreno-Romero et al. 2016). We could evaluate the parental H3K27me3 biases of 38 PEGs and 107 MEGs (Pignatta et al. 2014) after filtering out regions with H3 parental biases or without sufficient allele-specific reads. Consistent with findings from INTACT-ChIP data, we found that 31 of 38 PEGs were marked with Col-biased H3K27me3 peaks, while another three PEGs did not overlap with H3K27me3 peaks but exhibited Col biases on H3K27me3 profiles (Table S4). Only one of the 107 MEGs were associated with Ler-biased H3K27me3 peaks, and another 4 exhibited Ler biases in the H3K27me3 profile (Table S5). Additionally, 10 MEGs were marked by Col-biased H3K27me3 peaks (Table S5).
Discussion
In summary, CUT&RUN applied to FACS-purified endosperm nuclei enabled the examination of endosperm-specific H3K27me3 landscapes on maternally and paternally inherited genomes. CUT&RUN requires small amounts of starting nuclei and sequencing reads. It is also quick, taking only 1.5 days from collecting seeds for nuclei extraction to the completion of library preparation (Table 1). We showed here that CUT&RUN performed with about 20,000 nuclei and around 5 million sequencing reads is sufficient to profile the overall H3K27me3 landscape. To examine the parental-specific epigenome, we pooled four CUT&RUN replicates together in order to obtain greater allele-specific coverage. Even in this case, a total of just 80,000 nuclei were needed to generate around 3.5 million allele-specific reads (out of 26 million reads), which is a significant reduction from the 500,000 nuclei used for INTACT-ChIP-seq. Therefore, when combined with methods like FACS to isolate specific nuclei, CUT&RUN is particularly useful to profile the epigenome of specific types or groups of nuclei, especially those in low abundance. Additionally, because CUT&RUN does not require specific engineering of the nuclei, endosperm CUT&RUN can be applied to any plant species to profile the epigenome, as histones and histone modifications are highly conserved (Murr 2010; Talbert and Henikoff 2010) and a wide selection of histone-related antibodies are commercially available.
It is interesting that despite a large similarity, with 65% overlapping peaks, the endosperm H3K27me3 landscapes generated from CUT&RUN and INTACT-ChIP are quite distinct—each revealing about 30% unique peaks. By contrast, ChIP and CUT&RUN H3K27me3 profiles from leaves were almost identical. We found no evidence that H3K27me3 is enriched in pericentromeric heterochromatin in endosperm using CUT&RUN, unlike the results from INTACT-ChIP. This difference could originate from technical differences between the two methods, from potential biological differences between the sampled nuclei, or both. CUT&RUN utilizes a distinct methodology from INTACT-ChIP and relies on the DNA digestion activity of MNase when it is brought in close proximity to the protein of interest, perhaps making the technique more sensitive to local DNA–protein conformation, although it is not clear how this might affect the results. Additionally, the endosperm nuclei population sampled in our study is unlikely to have the exact same composition as that sampled in INTACT-ChIP, which could result in distinct H3K27me3 and H3 profiles. We purified only the 6C endosperm nuclei, excluding 3C endosperm nuclei. 6C nuclei, unless they represent endoreduplicated nuclei, should be in G2, and it is possible that cell cycle stage affects the distribution of H3K27me3 and H3. For INTACT-ChIP, endosperm nuclei were purified based on labeling by PHE1 promoter expression (Moreno-Romero et al. 2016, 2017). Though PHE1 is expressed in all endosperm nuclei (Moreno-Romero et al. 2016, 2017), the INTACT purification process might enrich for certain subtypes of endosperm nuclei with higher PHE1 expression. Thus, the endosperm nuclei populations used in INTACT-ChIP and our study could contain different proportions of micropylar, peripheral and chalazal endosperm nuclei, which might have distinct H3K27me3 and H3 landscapes, thus leading to different overall H3K27me3 peak profiles. Such biological differences are more likely to explain the differences in H3K27me3 peak identification around pericentromeric regions, since the H3K27me3 profiles between CUT&RUN and INTACT-ChIP differed in the opposite direction from that observed in the H3 profiles. In the future, it would be interesting to further separate sub-groups of endosperm nuclei and profile their epigenetic landscapes individually.
Experimental procedure
Plant material and crosses
Col-0 and Ler were grown in a growth chamber at 22 °C and 50% relative humidity under 16-h light and 8-h dark cycles. Col plants were emasculated and pollinated by Ler two days later. Seeds were removed from siliques four days after pollination (embryo at globular stage) for FACS sorting.
Antibodies used in this study
Anti-histone H3 (Sigma-Aldrich # H9289) in 1:100 final concentration (~ 15 µg/mL).
Anti-H3K27me3 (EMD Millipore # 07-449) in 1:100 final concentration (10 µg/mL).
Normal rabbit IgG (Cell Signaling Technology # 2729S) in 1:100 final concentration (10 µg/mL).
Solutions for FACS sorting and CUT&RUN
Protease Inhibitor 7x stock solution One cOmplete™ mini EDTA-free Protease Inhibitor Cocktail Tablets (Roche # 11836170001) in 1.5 ml dH2O. The stock solution can be stored at 2 to 8 °C for 1 to 2 weeks, or at least 12 weeks at − 15 to − 25 °C.
Nuclei extraction buffer Nuclei extraction buffer from Partec CyStain UV Precise P (Sysmex America # 05-5002) with 1/7 total volume of Protease Inhibitor 7x stock solution.
Nuclei staining buffer Staining buffer from Partec CyStain UV Precise P (Sysmex America # 05-5002) with 1/7 total volume of Protease Inhibitor 7x stock solution.
10x binding buffer 200 mM HEPES–KOH pH 7.9, 100 mM KCl, 10 mM CaCl2, 10 mM MnCl2 and 5 mM spermidine.
Binding buffer Diluted 10 times from 10x binding buffer, with 1/7 total volume of Protease Inhibitor 7x stock solution added.
Wash buffer 20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine and 0.1% BSA. Add one cOmplete™ Mini Protease Inhibitor Cocktail Tablets (Roche # 11836170001) to every 10 mL wash buffer. Keep on ice.
Wash buffer for formaldehyde-fixed nuclei Wash buffer with 1% Triton X-100 and 0.05% SDS. Keep on ice.
Blocking buffer Add EDTA solution (1 M, pH 8.0) to a final concentration of 2 mM to wash buffer.
Antibody buffer Aliquot 200 µL of wash buffer for each antibody and add antibody solution or serum to wash buffer to a final concentration of 1:100 or to the manufacturer’s recommended concentration. Make the antibody buffer right before it is needed.
2x stop buffer 200 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 µg/mL RNase A, 40 µg/mL glycogen and 10 pg/mL heterologous spike-in DNA. (We used the aliquot of yeast DNA from MNase-treated nucleosomes sent together with the protein A–MNase aliquot from the Henikoff laboratory.)
Experimental procedures
Notes
-
(A)
Detailed protocols for CUT&RUN and related tips can be found in Skene et al. (2018). To avoid redundancy, we only provide a step-by-step protocol for steps different from the Skene et al. protocol.
-
(B)
CUT&RUN performed on mammalian cells showed that flash-freezing of cells increased background cleavages (comments from Steven Henikoff), even though CUT&RUN on yeast nuclei extracted from flash-frozen yeast worked nicely (Skene and Henikoff 2017). We decided to avoid flash-freezing in the nuclei extraction step to maintain the integrity of DNA, but we have not tested the effect of flash-freezing plant samples on CUT&RUN.
-
(C)
Use cut tips when pipetting nuclei.
-
(D)
Quick spin before placing tubes with magnetic beads on magnetic stand.
-
(E)
For CUT&RUN using formaldehyde-fixed nuclei, it is important to use wash buffer with Triton X-100 and SDS to help the primary antibody and protein A–MNase access the nuclei through nuclear pores. Blocking buffer for formaldehyde-fixed nuclei also needs to be made from this wash buffer. The MNase active digestion time should be longer. In our experiment with formaldehyde-fixed nuclei, we activated MNase to digest for 1 h before stopping the reaction.
-
(F)
The DNA amount obtained from CUT&RUN is too little to quantify even with Qubit Fluorometric Quantitation. We only quality-checked the experiment after library preparation. Libraries should be able to be quantified by Qubit and exhibit a continuous distribution between 200 bp to 1200 bp using BioAnalyzer. CUT&RUN with the H3 antibody generates concentrated libraries with only 1000 nuclei and can be used as a positive control.
Extraction of nuclei from seeds*** for DAPI-based FACS sorting
-
1.
Prepare the proper amount of nuclei extraction buffer and nuclei staining buffer by adding Protease Inhibitor 7x stock solution. Wrap nuclei staining buffer with aluminum foil to protect it from light. Keep both buffers on ice.
-
2.
Aliquot 116.7 µL nuclei extraction buffer to each 1.5-ml microfuge tube on ice. For each tube, remove seeds from 7 to 8 siliques (4 DAP)* directly into buffer.
-
3.
Put a blue pestle into each tube, gently move the pestle up and down the solution to suspend the seeds, and then grind seeds manually in tube. Repeat suspension and grinding until the solution is homogenous and no big piece of whole tissue is visible.
-
4.
Add 800 µL nuclei staining buffer (with protease inhibitor added), pipette gently to mix.
-
5.
Filter the nuclei twice with CellTrics 30-µm filter (Partec # 04-0042-2316) for FACS sorting.
-
6.
Sort in flow cytometry machine (BD FACSAria)**. Use 1.5-mL tubes containing 50 µL PBS buffer to collect the sorted nuclei.
* From about 30 siliques, we usually collected around 50,000 6C nuclei.
** We sorted nuclei in a BD FACSAria SORP (special order research product) equipped with a 100 mW UV laser using the following parameters: Nozzle size, 85 microns; Sheath pressure: 45 psi; Droplet frequency: 45,000 drops/sec. Nuclei were gated based on signals from the DAPI channel.
*** For leaf samples, leaves were taken from 3-week-old plants and placed in a petri dish. 100 µL nuclei extraction buffer (with protease inhibitor added) was added to the surface of each leaf. The leaf was then chopped using a razor blade to fine pieces. 800 µL nuclei staining buffer (with protease inhibitor added) was then gently added to the chopped leaves, and nuclei were collected after filtering twice with CellTrics 30-µm filter for sorting.
Binding nuclei to magnetic beads
-
7.
(Optional) Fix sorted nuclei with formaldehyde if fixation of the protein–DNA interaction is necessary.
-
7.1.
Add 37% formaldehyde directly to the sorted nuclei to a final concentration of 1%. Vortex on low speed and incubate on ice for 8 min.
-
7.2.
Add glycine (2 M solution) to a final concentration of 0.125 mM. Vortex on low speed and incubate on ice for 5 min to stop cross-linking.
-
8.
Add 10x binding buffer to nuclei to a 1x final concentration.
-
9.
Gently vortex BioMag Plus concanavalin A-coated beads (Polysciences, Inc. #86057). Withdraw (10 x N) µL and add it to (40 x N) µL of binding buffer (N, sample number).
-
10.
After quick spin, place on a magnetic stand until the solution is clear (20 s to 1 min) and wash twice with (50 x N) µL binding buffer.
-
11.
Resuspend beads in (10 x N) µL binding buffer.
-
12.
Slowly add 10 µL beads to nuclei for each antibody treatment.
-
13.
Rotate 10 min at room temperature to allow binding.
Blocking nuclei
-
14.
After quick spin, place tubes on the magnetic stand to clear and get rid of the liquid.
-
15.
Add 500 µL blocking buffer to each sample and mix by inverting tubes. Make sure the blocking buffer has coated the entire inner surface of the tube.
-
16.
Rotate 5 min at room temperature.
Continue with step 10 to step 35 in the published CUT&RUN protocol (Skene et al.2018) to obtain DNA Use wash buffer instead of the digitonin buffer whenever mentioned in the Skene et al. protocol. For CUT&RUN with formaldehyde-fixed nuclei, skip step 34 and extract DNA using the following steps (modified based on Skene et al. 2018; Skene and Henikoff 2017).
DNA extraction for CUT&RUN with formaldehyde-fixed nuclei
-
(i)
After incubating at 37 °C for 10 min, add 2 µL 10% SDS (to 0.1%), 1.7 µL proteinase K (20 mg/ml) and 8.3 µL 5 M NaCl (to 0.2 M) to each sample. Vortex, incubate at 65 °C O/N to reverse cross-link.
-
(ii)
Add 200 µL buffered phenol–chloroform–isoamyl solution (25:24:1) and mix by vortexing at low speed.
-
(iii)
Transfer the solution to a phase-lock tube (Qiagen #129056), and centrifuge for 5 min at full speed at room temperature.
-
(iv)
Add 200 µL chloroform and invert about 10 times to mix. Centrifuge for 5 min at full speed at room temperature.
-
(v)
Remove aqueous layer to a fresh tube containing 2 µL of 2 mg/ml glycogen.
-
(vi)
Add 100 µL (half volume) Beckmann Agencourt AMPure XP beads and mix by pipetting up and down. Use cut tips because the mixture is very viscous.
-
(vii)
Let the mixture sit for 10 min at room temperature, and place on a magnetic stand to clear.
-
(viii)
Transfer the liquid to a fresh tube.
-
(ix)
Centrifuge the liquid for 5 min at full speed at 4 °C, transfer the liquid to a fresh tube to remove as many excess beads as possible.
-
(x)
Add 750 µL 100% ethanol, and mix by vortexing or tube inversion.
-
(xi)
Chill on ice for 5 to 10 min and centrifuge for 10 min at full speed at 4 °C.
-
(xii)
Pour off the liquid and drain on a paper towel.
-
(xiii)
Wash the pellet with 1 ml 100% ethanol and centrifuge for 1 min at full speed at 4 °C.
-
(xiv)
Carefully pipette out the liquid. Centrifuge for 1 min at full speed at room temperature. Pipette out the remaining liquid with 20-µL tips.
-
(xv)
Air-dry the pellet. When the pellet is dry, dissolve in 40 µL H2O.
Library preparation
Libraries were prepared using a protocol modified from the Henikoff protocol (http://blocks.fhcrc.org/steveh/papers/Codomo_Solexa_library_prep_protocol.docx). The purified DNA from CUT&RUN was first end-repaired, phosphorylated and adenylated in one reaction as follows. T4 DNA polymerase (NEB) was diluted 1:20 in 1x T4 DNA ligase buffer. Taq DNA polymerase (NEB) was diluted 1:20 in 1x Taq buffer. Each DNA sample (36.5 μL) was mixed with 5 μL 10x T4 DNA ligase buffer (NEB), 2.5 μL 10 mM dNTPs (KAPA), 1.25 μL 10 mM ATP (NEB), 3.13 μL 40% PEG4000, 0.63 μL T4 PNK (NEB), 0.5 μL diluted T4 DNA polymerase and 0.5 μL diluted Taq polymerase. The reaction was performed in a pre-cooled thermal cycler following the program: 12 °C for 15 min, 37 °C for 15 min and then 72 °C for 20 min. Samples were put on ice immediately, and adapter ligation was performed with addition of 5 µL of 1:50 diluted index adapter (Illumina TruSeq DNA PCR-free library prep kits FC-121-3001 and FC-121-3002), 55 µL 2x Rapid DNA ligase buffer (Enzymatics #B101L) and 5 µL T4 DNA ligase (Rapid; Enzymatics #L6030-HC-L) at 20 °C for 15 min. AMPure XP beads (Beckmann Agencourt) were immediately added to the sample after the ligation reaction in a ratio that excludes DNA fragments smaller than 150 bp and retains fragments equal or larger than 150 bp (around 0.33x, or 38 µL for 115 µL DNA for the batch of AMPure beads we used). The DNA–bead mixture was incubated at room temperature for 15 min and cleared in a magnetic stand, and the beads were washed twice with 200 μL 80% ethanol. After a quick spin, residual liquid was removed by pipette, and beads were air-dried for about 5 min, suspended with 53 μL nuclease-free dH2O and incubated at room temperature for 2 min. The beads were cleared from the supernatant on a magnetic stand and 50 μL of supernatant was retained. The DNA in supernatant was purified again with 1.1x (55 μL) AMPure XP beads with the same procedure described above and eluted in 20 μL nuclease-free dH2O. The purified adapter-ligated DNA was mixed with 10 μL 5x KAPA HiFi Fidelity Buffer, 1.5 μL 10 mM dNTPs, 1 μL KAPA HiFi HotStart DNA polymerase (KAPA #KK2502) and 0.5 μL each of 20 μM P5 and P7 primers. P5 and P7 primers were ordered from IDT with phosphorothioate bond between the 3′ A and G and PAGE purified (P5 Primer: 5′-AATGATACGGCGACCACCGA*G-3′. P7 Primer: 5′-CAAGCAGAAGACGGCATACGA*G-3′. * represents phosphorothioate bond). The mixed sample was PCR amplified to enrich the library following the program: 98 °C for 45 s, 98 °C for 15 s, 60 °C for 10 s, repeat the second and third steps 13 more times, and 72 °C for 1 min. The enriched library was purified again with 1.1x (55 μL) AMPure XP beads following the above-mentioned procedure. Purified libraries were examined by BioAnalyzer, and if adapter dimers (about 138-bp peak) or primer dimers (about 45-bp peak) were detected, the library was purified again with 1x AMPure XP beads following the same procedure described above.
Library sequencing
The published CUT&RUN protocol recommended paired-end sequencing (Skene et al. 2018). We performed 100 bp × 100 bp paired-end sequencing to maximize the number of allele-specific reads distinguished based on SNPs between Col and Ler. Twenty-four libraries were multiplexed into one sequencing lane in the flow cell of an Illumina HiSeq 2500s.
Data processing and analysis
Paired-end reads were trimmed and quality-filtered using trim_galore with –stringency 3 and quality cutoff -q 20. Both mates in a pair had to be at least 32 bp long after filtering and trimming. Trimmed qualified reads were aligned to TAIR10 genome using bowtie2 allowing zero mismatch in 22-bp seed region. Only reads that were uniquely mapped were retained for further analysis. PCR duplicated reads were removed using Picard “MarkDuplicates.jar”. Each replicate includes both H3K27m3 and H3 treatments, and was treated independently unless specified as merged. For the merged data, reads from all four replicates (two with unfixed nuclei and two with fixed nuclei) were merged together and the same alignment process was performed.
Epic (diffuse domain ChIP-Seq caller based on SICER, https://github.com/endrebak/epic; Zang et al. 2009) was used to call H3K27me3 peaks using H3K27me3 data and the corresponding H3 data as control with window size 150 bp and gap setting 2 (-w 150 -g 2). Since we were interested in histone modifications, we did not separate DNA fragments by sizes but rather analyzed all together. We used bamCoverage from deepTools 2.0 (Ramirez et al. 2016) to calculate read coverage per 150-bp bin using the reads per genomic content normalization option. The H3K27me3/H3 ratio in 150-bp bins was computed using bamCompute (deepTools 2.0; Ramirez et al. 2016). Heatmaps were generated using computeMatrix and plotHeatmap from deepTools 2.0 (Ramirez et al. 2016). Pearson correlation analysis was performed using multiBamSummary bins (–binSize 10000) and plotCorrelation (–corMethod pearson –skipZero; deepTools 2.0; Ramirez et al. 2016). In all calculations using deepTools, regions with no read coverage were skipped. Z-score was calculated using the following formula: \(\frac{{x - {\text{mean}}\left( x \right)}}{{{\text{standard}}\,{\text{deviation}}\left( x \right)}}\), where x is the log2 value of H3K27me3/H3 ratio in each 150-bp window.
Parental bias analysis
All uniquely mapped unduplicated reads from four CUT&RUN experiments were pooled together for higher allele-specific read coverage. The deduplicated aligned reads were assigned to a parent of origin by single-nucleotide polymorphisms (SNPs) between Col and Ler. multiBamSummary from deepTools 2.0 (Ramirez et al. 2016) was utilized to calculate allele-specific coverage of H3K27me3 and H3 in 150-bp windows. Windows with less than 20 H3 or H3K27me3 allele-specific reads (sum of Col- and Ler-specific reads) were filtered out. Col fraction of H3K27me3 and H3, respectively, in 150-bp bins were calculated (Col reads/(Col reads + Ler reads)). The top and bottom 10% of bins in the H3 Col fraction distribution were further filtered out. We identified allele-biased H3K27me3 regions based on the H3K27me3 Col fraction of each bin. The 90th percentile of H3 Col fraction (0.95) distribution is the cutoff for Col-biased H3K27me3 regions. Bins with H3K27me3 Col fraction higher than 0.95 were considered Col-biased. The 10th percentile of H3 Col fraction (0.68) distribution is the cutoff for Ler-biased H3K27me3 regions. Bins with H3K27me3 Col fraction lower than 0.68 were considered Ler-biased (Fig. 4a, b). Col- or Ler-biased H3K27m3 regions that overlapped with H3K27me3 peaks called by epic were considered Col- or Ler-biased H3K27me3 peaks (Fig. 4c).
References
Boisnard-Lorig C et al (2001) Dynamic analyses of the expression of the HISTONE:YFP fusion protein in arabidopsis show that syncytial endosperm is divided in mitotic domains. Plant Cell 13:495–509
Deal RB, Henikoff S (2010) A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell 18:1030–1040. https://doi.org/10.1016/j.devcel.2010.05.013
Gehring M (2013) Genomic imprinting: insights from plants. Annu Rev Genet 47:187–208. https://doi.org/10.1146/annurev-genet-110711-155527
Gehring M et al (2006) DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 124:495–506. https://doi.org/10.1016/j.cell.2005.12.034
Gehring M, Bubb KL, Henikoff S (2009) Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 324:1447–1451. https://doi.org/10.1126/science.1171609
Grossniklaus U, Vielle-Calzada JP, Hoeppner MA, Gagliano WB (1998) Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science 280:446–450
Hsieh TF, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL, Zilberman D (2009) Genome-wide demethylation of Arabidopsis endosperm. Science 324:1451–1454. https://doi.org/10.1126/science.1172417
Hsieh TF et al (2011) Regulation of imprinted gene expression in Arabidopsis endosperm. Proc Natl Acad Sci U S A 108:1755–1762. https://doi.org/10.1073/pnas.1019273108
Luo M, Bilodeau P, Koltunow A, Dennis ES, Peacock WJ, Chaudhury AM (1999) Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci U S A 96:296–301
Makarevich G, Villar CB, Erilova A, Kohler C (2008) Mechanism of PHERES1 imprinting in Arabidopsis. J Cell Sci 121:906–912. https://doi.org/10.1242/jcs.023077
Moreno-Romero J, Jiang H, Santos-Gonzalez J, Kohler C (2016) Parental epigenetic asymmetry of PRC2-mediated histone modifications in the Arabidopsis endosperm. EMBO J 35:1298–1311. https://doi.org/10.15252/embj.201593534
Moreno-Romero J, Santos-Gonzalez J, Hennig L, Kohler C (2017) Applying the INTACT method to purify endosperm nuclei and to generate parental-specific epigenome profiles. Nat Protocols 12:238–254. https://doi.org/10.1038/nprot.2016.167
Murr R (2010) Interplay between different epigenetic modifications and mechanisms. Adv Genet 70:101–141. https://doi.org/10.1016/B978-0-12-380866-0.60005-8
Ohad N, Margossian L, Hsu YC, Williams C, Repetti P, Fischer RL (1996) A mutation that allows endosperm development without fertilization. Proc Natl Acad Sci U S A 93:5319–5324
Park K et al (2016) DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc Natl Acad Sci U S A 113:15138–15143. https://doi.org/10.1073/pnas.1619047114
Pignatta D, Erdmann RM, Scheer E, Picard CL, Bell GW, Gehring M (2014) Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife 3:e03198. https://doi.org/10.7554/eLife.03198
Ramirez F et al (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44:W160–165. https://doi.org/10.1093/nar/gkw257
Skene PJ, Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6:e21856. https://doi.org/10.7554/elife.21856
Skene PJ, Henikoff JG, Henikoff S (2018) Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13:1006–1019. https://doi.org/10.1038/nprot.2018.015
Talbert PB, Henikoff S (2010) Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11:264–275. https://doi.org/10.1038/nrm2861
Wolff P et al (2011) High-resolution analysis of parent-of-origin allelic expression in the Arabidopsis endosperm. PLoS Genet 7:e1002126. https://doi.org/10.1371/journal.pgen.1002126
Xiao J et al (2017) Cis and trans determinants of epigenetic silencing by Polycomb repressive complex 2 in Arabidopsis. Nat Genet 49:1546–1552. https://doi.org/10.1038/ng.3937
Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W (2009) A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25:1952–1958. https://doi.org/10.1093/bioinformatics/btp340
Zhang X, Clarenz O, Cokus S, Bernatavichute YV, Pellegrini M, Goodrich J, Jacobsen SE (2007) Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol 5:e129. https://doi.org/10.1371/journal.pbio.0050129
Author contribution statement
XYZ and MG conceived and designed experiments. XYZ performed experiments and analyzed data. XYZ and MG wrote the manuscript.
Acknowledgements
We thank Steven Henikoff for generously providing protein A–MNase and suggestions on CUT&RUN, Arp Schnittger for providing a protocol for purifying endosperm nuclei using FACS, Morgan Moeglein, Colette Picard and P.R. Satyaki for optimizing the FACS protocol, Colette Picard for help and discussions on data analysis, and technical support from the Whitehead Institute FACS Facility and Genome Technology Core. This work was funded by NSF MCB award 1453459 to MG.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
High-throughput sequencing data have been deposited in NCBI GEO under accession GSE123602.
Additional information
Communicated by Dolf Weijers.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A contribution to the special issue ‘Cellular Omics Methods in Plant Reproduction Research’.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zheng, Xy., Gehring, M. Low-input chromatin profiling in Arabidopsis endosperm using CUT&RUN. Plant Reprod 32, 63–75 (2019). https://doi.org/10.1007/s00497-018-00358-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00497-018-00358-1