
Abstract
Acute and chronic liver injury results in hepatocyte death and turnover. If injury becomes chronic, the continuous cell death and turnover leads to chronic inflammation, fibrosis and ultimately cirrhosis and hepatocellular carcinoma. Controlling liver cell death both in acute injury, to rescue the liver from acute liver failure, and in chronic injury, to curb secondary inflammation and fibrosis, is of paramount importance as a therapeutic strategy. Both apoptosis and necrosis occur in the liver, but the occurrence of necroptosis in the liver and its contribution to liver disease is controversial. Necroptosis is a form of regulated necrosis which occurs in certain cell types when caspases (+/−cIAPs) are inhibited through the RIPK1-RIPK3 activation of MLKL. The occurrence of necroptosis in the liver has recently been examined in multiple liver injury models with conflicting results. The aim of this review is to summarize the published data with an emphasis on the controversies and remaining questions in the field.
Similar content being viewed by others


Facts
-
Hepatocytes do not express RIPK3 under basal conditions.
-
Blocking apoptosis in hepatocytes protects from cell death (with no switch to necroptosis).
-
Necrostatin-1 (RIPK1 kinase inhibitor) has been shown to protect against many models of acute liver injury such as acetaminophen toxicity.
Open Questions
-
What are the RIPK1 platform functions in hepatocytes versus RIPK1 kinase functions?
-
Does RIPK1 participate independent of the necrosome in hepatocyte death pathways?
-
Does necroptosis occur in non-parenchymal cells (NPCs)?
-
Can RIPK3 be induced in hepatocytes in chronic liver disease, and if so does this sensitize to necroptosis?
-
Does RIPK3-independent, but MLKL-dependent necrosis exist in hepatocytes in liver diseases?
The liver is a complicated vital organ responsible for metabolic processes, protein synthesis, and clearance of toxins and xenobiotics. The liver is the largest solid organ and has an unparalleled regenerative capacity, high vascular capacitance (receiving 30% of cardiac output) and is a site of immune-privilege.1,2 Hepatocytes are the main parenchymal cells making up 80% of liver mass, with the other epithelial cell type, the cholangiocytes lining the bile ducts, being the minority. The NPCs despite being high in number (40% of total cells) contribute to only 7% of liver volume because of their small size. These cells include the resident liver macrophages or Kupffer cells (KC), the liver sinusoidal endothelial cells (LSEC), the peri-sinusoidal hepatic stellate cells (HSC) formerly known as fat storing Ito cells, as well as liver lymphocytes particularly enriched in natural killer (NK) and natural killer T (NKT) cells. NPCs participate in normal physiologic liver functions and are also contributors to liver disease.
Liver injury is classified by duration into acute and chronic. Acute injury in its most extreme form results in acute liver failure (ALF), which is defined as an acute loss of hepatocyte mass (by necrosis or apoptosis) resulting in coagulopathy (elevated INR) and any degree of mental alteration (encephalopathy) in a patient without preexisting liver disease.3 Chronic liver injury is the result of long standing and ongoing liver damage and hepatitis from inflammation or intracellular stress responses (such as ER and mitochondrial stress). Common examples of chronic liver injury include steatohepatitis (alcoholic and non-alcoholic), autoimmune-hepatitis (AIH), primary biliary cholangitis, and chronic viral hepatitis (HBV, HCV). Acute and chronic liver injury results in hepatocyte death and turnover, which is commonly detected by an increase in the serum aminotransaminases (AST and ALT). If injury becomes chronic, the continuous cell death and turnover leads to chronic inflammation, activation of HSC into myofibroblasts and tissue repair responses, which result in fibrosis and ultimately cirrhosis and hepatocellular carcinoma. Controlling liver cell death both in acute injury to rescue the liver and in chronic injury to curb secondary inflammation and fibrosis is of paramount importance as a therapeutic strategy. When studying liver cell death, it is important to draw a distinction between acute and chronic liver disease models, since much of the controversy in the liver cell death field is because of these various contexts of injury under study. Different liver injury models differ in the type of signaling pathways activated, the role and the type of inflammatory cells involved, which ultimately dictate the cell death subroutine. For example, viral hepatitis results in cell death via death receptors (DR) and is executed mainly by adaptive cytotoxic T-cell activity resulting in liver cell apoptosis, whereas cell death from acetaminophen (APAP) hepatotoxicity, a hepatocyte intrinsic toxin, is largely necrotic and caspase and TNFR independent.4–7 This brings us to another point, and that is, although hepatocytes do express DR on their surface, not all liver cell death is DR-mediated and when comparing liver injury models one must take into account intrinsic death pathways activated by damage to the ER, mitochondria or nucleus, versus cytokine/DR ligand-activated DR signaling resulting in cell death.8
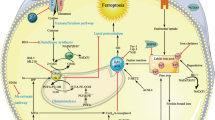
Necroptosis, is a mode of regulated cell death initiated by DR and occurs when caspases (+/−cIAPs) are inhibited. In certain cells when these conditions are met, RIPK1 and RIPK3 interact via their RIP homology interaction motif (RHIM) domains to form a complex recruiting the psudokinase mixed-lineage kinase domain-like (MLKL). MLKL is activated and phosphorylated by RIPK3 and subsequently translocates to the cell membrane resulting in its rupture (Figure 1).9 In the past few years, much attention has been directed towards the role of necroptosis in human disease. This has also led to an increasing number of studies to explore this cell death mode in the liver using various animal models of acute and chronic liver injury; however, a wide range of conflicting conclusions have been generated (Tables 1,2,3). Below we attempt to address the known facts, the intriguing controversies and the pressing questions in the field of liver cell death with a focus on the necroptosis pathway.

A simplified depiction of necroptosis induction by TNF. When TNF binds to its receptor (TNFR), complex 1 forms, which consists of the adaptor protein TRADD and ubiquitin ligases TRAF2 (also an adaptor), cIAPs, and LUBAC as well as the kinase, RIPK1. Ubiquitination of RIPK1 forms a platform to approximate key proteins leading to NFκB and MAPK activation and transcription of pro-survival and proi-nflammatory genes. Complex I internalization leads to the formation of the cytosolic complex II (not shown), which ultimately results in Caspase-8 activation culminating in apoptosis. When caspases +/− cIAPs are inhibited in certain cells, RIPK1 then forms a complex with RIPK3, which oligomerizes and recruits the pseudokinase, MLKL. RIPK3 phosphorylates MLKL that activates the protein leading to its translocation to cell membrane where it forms tetramers to permeate the lipid bilayer. TNF, tumor necrosis factor; TRADD, tumor necrosis factor receptor type 1-associated death domain; TRAF2, TNF receptor-associated factor 2, cIAP, cellular inhibitors of apoptosis; LUBAC, linear ubiquitin chain assembly complex; RIPK1, receptor interacting protein kinase 1; NFκB, nuclear factor κB; MAPK, mitogen-activated protein kinase; RIPK3, receptor Interacting protein kinase 3; MLKL, mixed-lineage kinase domain-like.
Do hepatocytes undergo necroptosis in models of liver injury?
Hepatocytes do not express RIPK3 under basal conditions
As briefly described above, necroptosis is a form of programmed cell death often mediated by TNFR1 and other DR engagement with their ligands. It occurs through the RIPK1-RIPK3-mediated activation of MLKL in certain cell types when there is inhibition of caspases +/−cIAPs.10 Activated MLKL (p-MLKL) forms a tetramer that translocates to the cell membrane where it executes necroptosis by a mechanism, which is still under debate.11–15 MLKL driven rupture of cell membrane is the necessary end point of necroptosis and the only known activator of MLKL at present is RIPK3. Therefore, RIPK3 and MLKL are necessary for the induction of necroptotic cell death.16
Although hepatocytes clearly express MLKL, the presence of RIPK3 in liver cells has been controversial.17 RIPK3, the third member of the receptor interacting protein kinase family was first described in 1999.18 RIPK3 mRNA distribution was examined using Northern blotting and its presence in the liver was not conclusive in the original report.18 Kasof et al.19 further dissected the role of RIPK3 in apoptosis in 2000 and reexamined its tissue distribution and failed to detect RIPK3 mRNA under basal conditions in the liver. RIPK3 protein is weakly present in whole liver homogenate (mixed cell types) but this in fact may be because of its presence in the NPC compartment. Much of the controversy lies with its induction during injury, which has been based mainly on western blotting and immunohistochemistry (IHC) of RIPK3 protein using commercially available non-specific polyclonal antibodies on injured liver tissue. Interestingly, in most reports, the area of RIPK3 staining corresponds to the area of necrotic or damaged liver. Injured liver tissue is highly immunoreactive, we have observed positive staining with non-specific normal rabbit and mouse IgG.17 Furthermore, we have observed positive RIPK3 staining in livers of RIPK3−/− mice using commercially available polyclonal antibodies, confirming the lack of specificity of these polyclonal antibodies.17 To determine if hepatocytes express RIPK3, we isolated primary mouse hepatocytes (PMH) and immediately measured RIPK3 mRNA and performed western blotting on freshly isolated PMH using a monoclonal antibody to RIPK3 provided to us by Dr. Newton of Genentech. We could not detect RIPK3 in hepatocytes under basal conditions nor after cell culture with APAP or TNF at different time points. Nor could we detect an increase in RIPK3 in PMH isolated from mice treated in vivo with APAP for 3 h.17 Recently others have confirmed this data, reporting much lower RIPK3 expression in whole liver homogenate compared with other organs such as spleen and intestine and undetectable RIPK3 in isolated mouse hepatocytes.20 Whether hepatocytes express RIPK3 or not is of critical importance since, as far as we know now, cells that do not express RIPK3 do not undergo necroptosis.21 Of course, theoretically in some unique circumstances it is possible that MLKL can be activated in a RIPK3-independent fashion and this has been suggested to occur in immune-mediated liver disease, see ConA section for more details.20,22
Blocking apoptosis in hepatocytes protects from cell death (with no switch to necroptosis).
Hepatocytes can die by necrosis and or apoptosis. In the liver, apoptosis is a common event in liver injury as hepatocytes robustly express DRs and many disease states have been shown to result in hepatocyte apoptosis, such as viral hepatitis (Councilman bodies), NASH (apoptotic bodies), alcoholic hepatitis, and autoimmune liver disease (adaptive immune-mediated).23,24 Direct DR activation under experimental conditions by TNF, FASL, TRAIL or DNA damage (ionizing radiation, cancer chemotherapeutic agents), or growth factor withdrawal have been shown to result in apoptotic cell death.23 Liver cells express DR (TNFR1, FAS, DR4/5), likely because of an evolutionary pressure to eradicate hepatotropic viruses. Indeed, during viral hepatitis T cells kill hepatocytes by DR-mediated apoptosis, which eliminates virally infected cells.24 Hepatocytes are type II cells and require the participation of the intrinsic mitochondrial apoptosis pathway for cell death completion.25,26 Only certain primary cells and cell lines such as L929 fibrosarcoma cells, Jurkat T cells, HT29 cells, MEF, keratinocytes express RIPK3 and have been shown to switch to an alternate form of cell death with DR signaling under conditions where caspases +/−cIAPs are inhibited.21 This has not been the case for all cells; for example, MCF-7, Hela cells and Hek293 cells do not undergo necroptosis because of a lack of RIPK3 expression.21 In fact, absence of RIPK3 in cells predicts whether they can undergo necroptosis.27 Under numerous apoptosis-inducing experimental conditions in hepatocytes we and others have observed a protection afforded by caspase inhibition without a switch to necroptosis.28–35 In addition, liver-specific ablation of caspase-8 has no effect on the normal liver and is in fact protective against Fas and TNF-mediated apoptosis.32,36 Therefore, the lack of a switch to a necroptotic form of cell death with caspase inhibition would argue against necroptosis occurring in liver cells. Necrosis of hepatocytes does occur in certain contexts but appears to be mediated by the mitochondrial permeability transition that can be considered a form of regulated necrosis. The MPT responds to ROS production and cyclophilin D controls pore opening.
Acute liver injury models in which necroptosis has been studied
The acetaminophen model
Acetaminophen (APAP) toxicity is a valuable model of liver cell death, hepatotoxicity and fulminant liver failure. The murine APAP model closely mimics human disease and is an excellent early translational model to study necrosis of hepatocytes. Once glucuronidation and sulfation pathways are saturated (in instances of overdose) APAP is metabolized to NAPQI via the Cyp450 system. NAPQI is highly electrophilic and covalently binds intracellular proteins and is thereby intrinsically toxic to hepatocytes. Inhibition or knockdown of many signaling molecules and MPT including components of the MAP Kinase pathway (MLK3, ASK1, JNK, Sab), GSK3b, PKCa, as well as cyclophilin D have been shown to protect against APAP.37–42 APAP-induced cell death has long been recognized to be necrotic and caspase and TNFR independent.4–7 It is a form of cell death which results in mitochondrial dysfunction and ROS generation, ATP depletion, MPT and necrotic oncosis. Given the involvement of signaling molecules, APAP hepatocyte death is a regulated form of cell death. However, as it pertains to necroptosis, it is important to point out that APAP cell death is independent of receptor signaling.7,43 Many investigators have evaluated the role of Necrostatin-1 (Nec-1) in APAP toxicity and have shown protection with the RIPK1 kinase inhibitor (Table 1).17,44–48 Yet, Nec-1 has off target effects and the true test of necroptosis is whether MLKL KO mice are protected against APAP. The effect of APAP on RIPK3 global knockout mice has been examined with varying conclusions. Two labs were unable to distinguish any protection against APAP toxicity at 24 h.17,45 However, Ramchandran et al.,45 observed protection against APAP (200–300 mg/kg) at 6 h but this protection did not extend to 24 h. We were unable to see any protection with APAP (300 mg/kg) using strain-matched controls at early or late times points.17 Deutsch et al.46 reported a robust protection against APAP (700 mg/kg) with a 75% survival advantage compared with C57BL/6j controls. In our hands 700 mg/kg of APAP results in 100% mortality in all WT and transgenic mice models (C57BL/6n).
The discrepancy in the reports may be in part because of experimental variations (such as APAP preparation methods) as well as strain matching. The Genentech generated RIPK3−/− mice are on an n sub-strain (personal communication with Genentech), whereas WT control mice used in the Ramachandran paper were j sub-strain and the C57BL/6 mice in the Deutsche paper were purchased from Jackson (likely j sub-strain unless otherwise requested). C57BL/6 substrains n and j have different susceptibility to APAP liver injury.49 Finally, given the controversy with the RIPK3 knockout mice and the fact that MLKL is indispensable for the execution of necroptosis, we examined the effect of APAP 300 mg/kg on MLKL−/− mice (compared with j sub-strain-matched controls) and found no attenuation of injury.17 This demonstrates that cell death in the APAP model is MLKL independent and thus most likely not necroptosis.17 Gunther et al.20 have independently obtained similar results, MLKL−/− mice were not protected against APAP compared with WT controls (j sub-strain).20 Although these results seem confirmatory, more rigorous studies using littermate controls and inducible liver cell-specific knockout are needed for conclusive results.22 Furthermore, we could find no change in RIPK3 expression in the short time course APAP toxicity (up to 24 h) in vitro or in vivo.
Immune activation models: concanavalin A (ConA) and α-galactosylceramide (α-GalCer)
The plant lectin concanavalin A (ConA) is widely used to induce ‘immune-mediated’ acute liver injury in mice. It is often used as a model of hepatitis as the hepatocellular injury in ConA is primarily initiated by the direct activation of T cells, particularly CD4+ T cells and NKT cells and targeting of hepatocytes by the immune system in an antigen TCR-independent fashion.50–54 Subsequent activation of macrophages/KC and recruitment of leukocytes into the liver significantly contributes to the liver injury.55,56 The mode of cell death in ConA is largely necrotic, although some reports have observed DNA fragmentation and apoptotic bodies as early as 5 h after ConA injection, suggesting that apoptosis of hepatocytes occurs early in the injury phase followed by massive cell lysis and necrosis in the later hours.57,58 TNF is presumed to be the major cytokine activating hepatocyte DR as interference with TNF (treatment with anti-TNF antibodies) has been shown to be protective against ConA-induced hepatitis and hepatocellular death.57–59 TNF is directly capable of inducing hepatocyte apoptosis via TNF-receptor signaling inducing caspase-8 activation and involvement of the mitochondrial intrinsic death pathway, leading to mitochondrial cytochrome C release and caspase-3 activation. However, as TNF signaling also activates NF-κB, which in turn switches on pro-survival gene expression such as cFLIP, BclXL among others, in order for TNF to mediate hepatocyte death, the deactivation or inhibition of the NF-κB pathway is necessary. However, this does not seem to be the case with ConA as the lectin alone can result in hepatitis and cell death without the need for transcriptional or translational inhibition.57 In addition to TNF, other major cytokines such as IFN-γ, IL4, IL6 and other DR pathways (Fas, TRAIL) have also been implicated in ConA hepatotoxicity, although to a lesser extent than TNF.60–65 The ConA model is very complex. In addition to immune cells, LSECs have also been suggested to have a pivotal role in the injury by mediating a hypercoagulative state in the liver.66
In spite of the immune-mediated features of ConA toxicity, such as the requirement for TNF and IFN-γ signaling, liver cell death appears to be necrosis morphologically and not apoptosis. This is further supported by the fact that no significant activation of caspase activity has been observed with ConA and no protection from liver injury is afforded by caspase inhibition.67,68 Given the involvement of the TNF DR pathway and the late appearance of necrotic cell death in ConA model, multiple laboratories have investigated whether necroptosis and the RIPKs contribute to ConA-induced hepatitis (Table 2). Jouan-Lanhouet et al.69 examined the effect of necrostatin-1 (125 µg) administration 15 min before on ConA (20 mg/kg, retro-orbital injection) induced hepatitis on liver damage using female C57BL6 mice and reported a decrease of about 50% in ALT and histologic injury versus PBS pre-treatment. Nec-1 is dissolved in DMSO, which is known to be hepatocyte protective, may have some confounding effects especially if not controlled for. They demonstrated that ConA-activated PARP-1 in the liver of WT mice and that the PARP-1 inhibitor, PJ-34, protected to the same extent as Nec-1. Nec-1 not only resulted in less hepatotoxicity as measured by ALT but also decreased PARP-1 activation, suggesting that PARP-1 may be downstream of RIPK1 kinase activation in ConA.69 Recently, the same group published a follow up paper, this time using 12 mg/kg of ConA and the same pre-treatment protocol with Nec-1 125 µg, 15 min before ConA and they concluded that protection by RIPK1 inhibitor and PARP inhibitor was because of lower levels of IL-33.70 Zhou et al.,71 performed similar experiments on male C57BL/6 mice. They administered Nec-1 (1.8 mg/kg) or DMSO via tail vein 1 hour before ConA (20 mg/kg) and observed less AST and ALT as well as a survival benefit with Nec-1 pre-treatment (30% versus 7% mortality). Deutsch et al. while studying necroptosis in the ConA model observed that although RIPK3−/− mice were mildly protected as measured by ALT compared with WT C57BL/6, this did not translate to a survival advantage. However, interestingly, simultaneous treatment of WT mice with Nec-1 (1.6 mg/kg) and ConA (20 mg/kg) compared with ConA (20 mg/kg) alone exacerbated liver injury, more than doubling the ALT and resulting in 50% mortality from Nec-1 treatment.46
Deutsche et al. investigated the mode of cell death in ConA using a global knockout of RIPK3. Even though they interpret RIPK3 deletion as being protective, there was only a modest (<10%) decrease in transaminases.46 These minor differences in in vivo experiments using possibly non-strain-matched mice that are not littermates should be interpreted with caution. It is interesting that Deutsche and colleagues observed a robust aggravation of injury with Nec-1 and ConA co-treatment compared with ConA alone in contrast to findings by Zhou et al.,71 Jouan-Lanhouet et al.69 and Arshad et al.70 The protection reported with Nec-1 should also be further scrutinized given the possible off target effects of Nec-1.72 Interestingly, Weinlich et al.73 have also examined the effect of ConA on RIPK3−/− mice compared with WT littermates and noted that the knockout animals displayed the same level of liver damage as control littermates, strongly arguing against RIPK3-dependent necroptosis in ConA liver injury. Recently Gunther et al.20 have reported protection against ConA (25 mg/kg) in global MLKL−/− mice. Interestingly, whereas Nec-1 s (400 µg i.p.) was also protective in their experiments, RIPK3 knockout was not. In addition, they noted an increase in both RIPK1 and MLKL protein after ConA, whereas RIPK3 protein levels, which are very weak in whole liver and absent in hepatocytes did not change or increase after ConA liver injury. It is unclear in these studies how MLKL is activated in the absence of RIPK3, but an unidentified kinase downstream of RIPK1 is suggested to contribute to this non-canonical necroptosis pathway which seems to be MLKL-dependent.20,22 Investigation of conditional and liver-specific knockout models is still warranted.
Another model of immune cell activation is α-galactosylceramide (α-GalCer), a specific activator for invariant NKT cells resulting in immune-mediated liver injury and induction of regulatory and inflammatory cytokines. In WT mice α-GalCer activation of NKT cells results in increased TNF, IFN-γ, IL4, and subsequent activation of innate immune cells in the liver, causing a mild to moderate liver injury (ALT ~300–1000 IU) at the peak time of 16–24 h. This immune-mediated model is quite similar to the ConA model, but relatively less severe and initiated by specific activation of NKT cells, which are uniquely abundant in the liver. Kang et al.28 have reported that both α-GalCer injury and ConA-induced liver damage is attenuated in global RIPK3−/− mice (ALT 200) accompanied by less TNF and IFNγ mRNA expression. To confirm that the RIPK3 deletion in NKT cells was attenuating the injury, they performed bone marrow (BM) transplants between RIPK3−/− and WT mice to generate chimeric animals. They showed that RIPK3 in BM cells was essential for liver injury to occur as ConA-induced liver injury was significantly attenuated in the WT animals that had RIPK3−/− BM. They provided evidence showing that the reduced liver injury in RIPK3−/− mice is not related to necroptosis of hepatocytes, and most likely because of an impairment of production of cytokines (TNF and IFN-γ) by RIPK3-deficient NKT cells in a necroptosis-independent fashion. Thus, these findings are very interesting but do not support the hypothesis of necroptosis occurring in hepatocytes as the RIPK3 deletion in NKT cells seems to be the limiting factor.28 We have also been interested in the α-GalCer model of liver injury and have examined the role of RIPK3 and MLKL deletion as well as RIPK1 knockdown via antisense oligonucleotide in NKT-mediated immune injury from α-GalCer. Contrary to the findings by Kang et al., we found no difference in liver injury between global RIPK3−/−, MLKL−/− mice and strain-matched controls.74 However, we found that knockdown of RIPK1 markedly exacerbated α-GalCer-mediated liver injury and induced lethality as early as 6 h after treatment. Unexpectedly, despite the massive hepatocyte death and the effective RIPK1 knockdown (>95%) the exacerbated liver injury was not because of an impaired hepatic NF-κB activation. As we observed no difference with RIPK3−/− or MLKL−/− mice, we concluded that necroptosis was not involved. We observed increased TUNEL staining and caspase-3 activation in the RIPK1 knockdown animals with severe liver injury and we suspected the mode of death was in fact apoptosis. On further investigation, we observed that the exacerbation of liver injury with RIPK1 knockdown was because of TNF-and caspase-dependent massive apoptosis of hepatocytes as neutralization TNF or inhibition of caspase by pan-caspase inhibitor zVAD.fmk conferred an almost complete protection.74 Interestingly, ZVAD protection was not accompanied by a switch to necroptosis.
Chronic liver models
Alcoholic and non-alcoholic steatohepatitis (ASH and NASH) along with models of viral hepatitis are the most commonly studied chronic liver injury models. Studies of mechanisms of hepatocyte cell death in viral hepatitis have been somewhat restricted by the limited availability of in vivo animal models. However, studies of hepatitis C in cell culture have to a large extent concluded the mode of cell death as apoptotic.75–77 One study has reported an additive protection with in vitro utilization of Nec-1 to the pan-caspase inhibitor QVD, although this was examined on Huh7 cells, and not primary hepatocytes and there was no mechanistic exploration as to how Nec-1 was exerting its effects.78
Non-alcoholic fatty liver disease and steatohepatitis (NAFLD/NASH) are a leading cause of liver disease and obesity related mortality is the US with increasing prevalence worldwide.79 Chronic alcohol use also results in steatohepatitis and is an important cause of cirrhosis worldwide. Long standing steatohepatitis results in inflammation, cell death, fibrosis, leading to cirrhosis and hepatocellular carcinoma. Therefore, therapeutic strategies other than weight loss and abstinence from alcohol are sorely needed.79 So far, the mode of cell death in NASH is presumed to be apoptosis as interfering with apoptosis, caspase inhibition and liver-specific caspase-8 knockout greatly protect.80 However, studying animal models of steatohepatitis has proven to be challenging. Unlike the APAP model which very closely mimics human pathology, it is difficult to study alcoholic steatohepatitis (ASH) and NASH by replicating robust steatohepatitis in mice using alcohol or a high fat diet (HFD) alone. Mice do not develop the same degree of inflammation and fibrosis as humans which may be a function of the duration and intensity of alcohol or HFD feeding or normal physiologic metabolic differences.81 In humans, fatty liver and NASH are accompanied by visceral adiposity, insulin resistance, type II diabetes and metabolic syndrome, which has proven somewhat difficult to recapitulate in animal models. The same is true for alcohol, existing rodent models of alcoholic liver disease result in less liver injury from alcohol (lower inflammation, ALT and bilirubin) in mice and rats compared with the effect of alcohol on human liver.82 Nevertheless, using the available animal models the role of necroptosis has been investigated in ASH and NASH (Table 3).
Roychowdhury et al.83 examined the effects of RIPK3 in mice fed alcohol. They reported an increase in RIPK3 expression by IHC using a commercially available polyclonal antibody with the Lieber de Carli model of free access to alcohol as well as chronic binge feeding. The RIPK3 staining was more prominent around the central vein. In addition, the authors report RIPK3 induction in human liver biopsy samples of patients with alcoholic liver disease using IHC. In RIPK3−/− mice (back-crossed to j sub-strain) they reported a modest attenuation of liver injury compared with WT C57BL6/j sub-strain mice (ALT in WT of 40, AST 70 down to 20 and 40, respectively, for RIPK3−/−). They also reported less liver triglycerides, TNF and liver inflammatory cell numbers by IHC. Wang and colleagues used the Gao-binge model of alcohol feeding and reported an increased expression of RIPK3 using a (polyclonal RIPK3 antibody) with alcohol and protection from global RIPK3 deletion (ALT and steatosis) but no difference in hepatitis and neutrophil infiltration in the RIPK3−/− mice. They could not detect a transcriptional change (mRNA) in RIPK3 and attributed the increased protein levels of RIPK3 to impaired hepatic proteasome function because of less protein turnover resulting from alcohol feeding.84,85 Roychowdhury et al.86 have recently published a follow up paper to using a HFD model and comparing RIPK3−/− and WT controls. They find no protection against HFD-induced steatohepatitis with global RIPK3 knockout. In fact, they report worsening of injury compared with controls. The RIPK3−/− animals had basal insulin resistance, which significantly worsened on a HFD leading to steatosis, inflammation and fibrosis. It is interesting that RIPK3 knockout had diametrically opposite results in the alcohol model and HFD model experiments performed on the same mice by the same group. Even more intriguing is that in both instances the authors report increased RIPK3 in the liver by immunostaining.83,86 The chow-fed wild-type mice in the present study expressed no basal RIPK3 or p-MLKL, but after HFD feeding showed increased expression of both proteins by IHC. However, despite the high expression of these necroptosis proteins the mice had very little injury. Therefore it is highly unlikely for necroptosis to be activated in this model.86
Two groups have investigated the role of RIPK3 in liver injury, using the methionine choline-deficient model, which is used as a NASH model.87,88 Gautheron et al.88 demonstrate an increase in RIPK3 expression using IHC and western blotting (polyclonal antibody). They also reported that liver injury is worsened in the animals by caspase-8 liver-specific knockout. Interestingly, in the liver-specific caspase-8 KO increased hepatic RIPK3 expression was observed even in the control diet, which may be because of compensatory mechanisms that occur in embryonic knockout animal models, making them difficult to interpret. Alfonso et al.87 have used IHC on human livers with various diseases and have demonstrated an increase in RIPK3 staining in liver sections from humans with NASH, ASH and hepatitis B and C. One could interpret this one of two ways, either all of these human liver diseases result in RIPK3 upregulation and as the authors suggest an involvement of necroptosis, or the antibody is non-specifically binding injured liver tissue. The authors offer no explanation as to how caspases are inhibited in these disease states for necroptosis to ensue. In mice using the high fat choline-deficient and methionine deficient models, Alfonso and colleagues demonstrated an increase in RIPK3 and p-MLKL in the insoluble liver fraction and a protection from RIPK3 knockout. Curiously, although TNF (plus transcriptional or translation arrest) is known to induce apoptosis in hepatocytes,8,89–91 the authors were unable to prevent cell death from TNF/CHX by caspase inhibition. Both of these studies used the methionine and or choline-deficient models which are not ideal models of NASH because of a lack of insulin resistance and weight gain in the animals.92,93 Furthermore, the technical limitations and non-specific binding of polyclonal antibodies used in IHC of the liver, in our opinion, limits the conclusions one can reach from these descriptive studies.17
Regardless, assuming the RIPK3 induction in hepatocytes is indeed robust and relevant in chronic disease models, we are left with the dilemma of understanding how physiologic necroptosis occurs in the presence of caspases, as all models of necroptosis require the genetic deletion or inhibition of caspases +/− IAPs.21,92 One must also take into account that these models are global RIPK3 knockouts and it is not clear whether the observed protection from lack of RIPK3 is because of a hepatocyte intrinsic mechanisms or an effect on the immune system. In certain disease models, which exhibit hemorrhage or severe inflammation in the liver, the inflammatory cells and RBC infiltration to the liver could theoretically increase total RIPK3 levels because of the mixed cell population. Nevertheless, although we do not believe that RIPK3 is upregulated in in acute liver injury, we recognize that context is important and it is possible that in chronic models of liver injury RIPK3 is indeed induced in hepatocytes. Whether this has any relevance or correlation with necroptosis in the absence of caspase inhibition remains to be determined. Furthermore, one has to consider cell death-independent functions of RIPK3 expression in hepatocytes.
Unclassified Drug-Induced Liver Injury
Wang et al.15 while testing a phospho-MLKL antibody as a specific necroptosis marker had reported positive staining in liver biopsy samples from patients with drug-induced liver injury (DILI). The authors however do not specify what kind of DILI samples they used and what drug or toxicant was the culprit. Drug-induced liver injury is a complicated clinical diagnosis with a wide range of presentations and a lack of objective diagnostic test, making it largely a diagnosis of exclusion.94 In the study by Wang et al.15 no clinical data or patient characteristics, pattern of injury (hepatocellular or cholestatic) or identification of suspected drugs was provided. It is extremely important to emphasize that although some drugs such as acetaminophen are direct hepatotoxins, most drug toxicities are idiosyncratic and HLA/adaptive immune-mediated.95 DILI presents in a diverse and heterogeneous fashion, some drugs target hepatocytes and cause a hepatocellular pattern of injury, whereas others result in cholestasis. Some drugs result in hepatocyte necrosis, some activate immune cells to target DR-mediated apoptotic cell death, whereas other drugs result in vanishing bile ducts and are mainly toxic to cholangiocytes. The authors report patchy p-MLKL staining of dead hepatocytes around the central vein area of the 14 reported ‘DILI patients’.15 This is an intriguing descriptive finding. However, there is not a mechanistic explanation. Furthermore, if this implicates necroptosis activation, one would have to assume that in these 14 patients, caspase activation was somehow inhibited or defective for the hepatocytes to not die of apoptosis as they are very prone to do and instead default to necroptosis. This scenario has yet to be shown in physiologic conditions.
Remaining questions
Does RIPK1 participate independent of the necrosome in hepatocyte death pathways?
Multiple groups have studied the role of Nec-1 in models of acute liver injury in particular APAP, and have observed protection. Given the off target effects of Nec-1, we examined the effect of RIPK1 knockdown using an IONIS antisense oligonucleotide which concentrates in the liver and knocks down RIPK1 with about 95% efficiency. As RIPK3−/− and MLKL−/− mice were not protected but knockdown of RIPK1 in these mice afforded partial protection against APAP necrosis, we believe RIPK1 may have a novel and independent function in hepatocyte death, which appears to be upstream of JNK. One of our most striking observations in the past few years was that knockdown of RIPK1 protected against hepatocyte death in the APAP toxicity model while worsening injury from immune-mediated liver injury caused by α-GalCer. These two models are vastly different. The former is a cell intrinsic toxin to hepatocytes, which mediates a cyclophilin D and MPT-dependent necrotic form of cell death and the latter a DR-mediated apoptosis of hepatocytes induced by activated-NKT cells. It should not be surprising that RIPK1, which is a key molecule in inflammatory pathways, functions both as a survival protein and a death-inducing protein in a context-dependent manner.9,96,97 Furthermore, RIPK1 has been suggested as a participant in ER stress-induced apoptosis,98 and it is likely that RIPK1 has additional functions beyond mediating apoptosis and necroptosis.9 We should note that although we did observe a protection against APAP with RIPK1 knockdown, others have observed no protection with embryonic RIPK1 liver-specific knockout.99 Although the antisense may have off target effects, there are also issues with embryonic knockout, namely the capacity of living organisms to compensate in unexpected ways for the loss of vital proteins and alterations in normal homeostasis. It remains to be seen if adult inducible liver-specific knockout of RIPK1 protects against any of the acute and chronic liver injury models and hepatocyte death.
Does necroptosis occur in NPCs?
As discussed in the introduction, hepatocytes are not the only liver cell type. In fact, the liver has a rich and diverse microenvironment harboring multiple types of specialized cells. Hepatocytes function mainly to regulate metabolism, detoxify xenobiotics, synthesize critical circulating proteins, and generate and secrete bile acids. The NPCs, including the LSECs, KC, HSC, NK, NKT, are in constant cross talk with hepatocytes and each other. We have observed a robust expression of RIPK3 and MLKL in isolated LSECs, KCs and liver leukocytes.17 As NPC participate in normal liver functions and have all the required machinery for the execution of necropoptosis, it is intriguing to hypothesize that NPC necropoptosis may contribute to some forms of liver disease.
Summary of Key Points
-
RIPK3 is not expressed under basal conditions in hepatocytes.
-
As MLKL is expressed in hepatocytes there may yet be a RIPK3-independent mechanism for activation of MLKL and necroptosis in these cells.
-
RIPK3 may be induced in certain disease conditions of the liver, but if so, necroptosis-independent pathways must always be considered as well.
-
Preventing apoptosis in hepatocytes after DR engagement does not result in a switch to necroptosis; rather it prevents cell death.
-
Human liver disease conditions in which caspases are inhibited (a requirement for necroptosis) have yet to be reported.
-
More conclusive littermate controlled studies are needed using cell-specific inducible knockouts of MLKL, and the RIPKs and valid antisera and reagents that are specific and suitable for immunoblotting to resolve controversies and uncertainties.
References
Knolle PA, Gerken G . Local control of the immune response in the liver. Immunol Rev 2000; 174: 21–34.
Lautt WW . Hepatic Circulation: Physiology and Pathophysiology. Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2009.
WML Lee, Anne M, Stravitz RT (eds). AASLD Position Paper: The Management of Acute Liver Failure. Update 2011.
Jaeschke H, Cover C, Bajt ML . Role of caspases in acetaminophen-induced liver injury. Life Sci 2006; 78: 1670–1676.
Jaeschke H, Williams CD, Farhood A . No evidence for caspase-dependent apoptosis in acetaminophen hepatotoxicity. Hepatology 2011; 53: 718–719.
McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H . The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest 2012; 122: 1574–1583.
Boess F, Bopst M, Althaus R, Polsky S, Cohen SD, Eugster HP et al. Acetaminophen hepatotoxicity in tumor necrosis factor/lymphotoxin-alpha gene knockout mice. Hepatology 1998; 27: 1021–1029.
Malhi H, Guicciardi ME, Gores GJ . Hepatocyte death: a clear and present danger. Physiol Rev 2010; 90: 1165–1194.
Newton K . RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol 2015; 25: 347–353.
Silke J, Rickard JA, Gerlic M . The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol 2015; 16: 689–697.
Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell 2016; 61: 589–601.
Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013; 39: 443–453.
Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 2014; 111: 15072–15077.
Chen X, Li W, Ren J, Huang D, He WT, Song Y et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 2014; 24: 105–121.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54: 133–146.
Murphy JM, Vince JE . Post-translational control of RIPK3 and MLKL mediated necroptotic cell death. F1000Res 2015; 4: F1000.
Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015; 62: 1847–1857.
Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM . RIP3, a novel apoptosis-inducing kinase. J Biol Chem 1999; 274: 16871–16875.
Kasof GM, Prosser JC, Liu D, Lorenzi MV, Gomes BC . The RIP-like kinase, RIP3, induces apoptosis and NF-kappaB nuclear translocation and localizes to mitochondria. FEBS Lett 2000; 473: 285–291.
Gunther C, He G, Kremer AE, Murphy JM, Petrie EJ, Amann K et al. MLKL mediates programmed hepatocellular necrosis independent of RIPK3 during hepatitis. J Clin Invest 2016; 4346–4360.
Degterev A, Zhou W, Maki JL, Yuan J . Assays for necroptosis and activity of RIP kinases. Methods Enzymol 2014; 545: 1–33.
Dara L, Liu Z, Kaplowitz N . A murder mystery in the liver: who done it and how? J Clin Invest 2016; 126: 4068–4071.
Ding WX, Yin XM . Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med 2004; 8: 445–454.
Yoon JH, Gores GJ . Death receptor-mediated apoptosis and the liver. J Hepatol 2002; 37: 400–410.
Zhao Y, Ding WX, Qian T, Watkins S, Lemasters JJ, Yin XM . Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology 2003; 125: 854–867.
Bradham CA, Qian T, Streetz K, Trautwein C, Brenner DA, Lemasters JJ . The mitochondrial permeability transition is required for tumor necrosis factor alpha-mediated apoptosis and cytochrome c release. Mol Cell Biol 1998; 18: 6353–6364.
Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J et al. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis 2015; 6: e1884.
Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun 2015; 6: 8371.
Thapaliya S, Wree A, Povero D, Inzaugarat ME, Berk M, Dixon L et al. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig Dis Sci 2014; 59: 1197–1206.
Hatting M, Zhao G, Schumacher F, Sellge G, Al Masaoudi M, Gabetaler N et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology 2013; 57: 2189–2201.
Kaufmann T, Jost PJ, Pellegrini M, Puthalakath H, Gugasyan R, Gerondakis S et al. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity 2009; 30: 56–66.
Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol 2004; 173: 2976–2984.
Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009; 50: 1421–1430.
Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ . The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther 2004; 308: 1191–1196.
Zender L, Hutker S, Liedtke C, Tillmann HL, Zender S, Mundt B et al. Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc Natl Acad Sci USA 2003; 100: 7797–7802.
Liedtke C, Bangen JM, Freimuth J, Beraza N, Lambertz D, Cubero FJ et al. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology 2011; 141: 2176–2187.
Han D, Dara L, Win S, Than TA, Yuan L, Abbasi SQ et al. Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends Pharmacol Sci 2013; 34: 243–253.
Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N . c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006; 131: 165–178.
Win S, Than TA, Han D, Petrovic LM, Kaplowitz N . c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem 2011; 286: 35071–35078.
LoGuidice A, Boelsterli UA . Acetaminophen overdose-induced liver injury in mice is mediated by peroxynitrite independently of the cyclophilin D-regulated permeability transition. Hepatology 2011; 54: 969–978.
Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A et al. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology 2008; 135: 1311–1321.
Sharma M, Gadang V, Jaeschke A . Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol 2012; 82: 1001–1007.
Williams CD, McGill MR, Farhood A, Jaeschke H . Fas receptor-deficient lpr mice are protected against acetaminophen hepatotoxicity due to higher glutathione synthesis and enhanced detoxification of oxidant stress. Food Chem Toxicol 2013; 58: 228–235.
An J, Mehrhof F, Harms C, Lattig-Tunnemann G, Lee SL, Endres M et al. ARC is a novel therapeutic approach against acetaminophen-induced hepatocellular necrosis. J Hepatol 2013; 58: 297–305.
Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H . The receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013; 58: 2099–2108.
Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis 2015; 6: e1759.
Zhang YF, He W, Zhang C, Liu XJ, Lu Y, Wang H et al. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett 2014; 225: 445–453.
Takemoto K, Hatano E, Iwaisako K, Takeiri M, Noma N, Ohmae S et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Biol 2014; 4: 777–787.
Bourdi M, Davies JS, Pohl LR . Mispairing C57BL/6 substrains of genetically engineered mice and wild-type controls can lead to confounding results as it did in studies of JNK2 in acetaminophen and concanavalin A liver injury. Chem Res Toxicol 2011; 24: 794–796.
Heymann F, Hamesch K, Weiskirchen R, Tacke F . The concanavalin A model of acute hepatitis in mice. Lab Anim 2015; 49: 12–20.
Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K . Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci USA 2000; 97: 5498–5503.
Kaneko Y, Harada M, Kawano T, Yamashita M, Shibata Y, Gejyo F et al. Augmentation of Valpha14 NKT cell-mediated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in the development of concanavalin A-induced hepatitis. J Exp Med 2000; 191: 105–114.
Toyabe S, Seki S, Iiai T, Takeda K, Shirai K, Watanabe H et al. Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J Immunol 1997; 159: 1537–1542.
Tiegs G, Hentschel J, Wendel A A . T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest 1992; 90: 196–203.
Bonder CS, Ajuebor MN, Zbytnuik LD, Kubes P, Swain MG . Essential role for neutrophil recruitment to the liver in concanavalin A-induced hepatitis. J Immunol 2004; 172: 45–53.
Schumann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N et al. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol 2000; 157: 1671–1683.
Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A . Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol 1995; 146: 1220–1234.
Mizuhara H, O'Neill E, Seki N, Ogawa T, Kusunoki C, Otsuka K et al. T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med 1994; 179: 1529–1537.
Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP et al. Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology 1998; 114: 1035–1045.
Wang HX, Liu M, Weng SY, Li JJ, Xie C, He HL et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J Gastroenterol 2012; 18: 119–125.
Takahashi K, Murakami M, Kikuchi H, Oshima Y, Kubohara Y . Derivatives of Dictyostelium differentiation-inducing factors promote mitogen-activated IL-2 production via AP-1 in Jurkat cells. Life Sci 2011; 88: 480–485.
Miller ML, Sun Y, Fu YX . Cutting edge: B and T lymphocyte attenuator signaling on NKT cells inhibits cytokine release and tissue injury in early immune responses. J Immunol 2009; 183: 32–36.
Gove ME, Rhodes DH, Pini M, van Baal JW, Sennello JA, Fayad R et al. Role of leptin receptor-induced STAT3 signaling in modulation of intestinal and hepatic inflammation in mice. J Leukoc Biol 2009; 85: 491–496.
Zheng SJ, Wang P, Tsabary G, Chen YH . Critical roles of TRAIL in hepatic cell death and hepatic inflammation. J Clin Invest 2004; 113: 58–64.
Seino K, Kayagaki N, Takeda K, Fukao K, Okumura K, Yagita H . Contribution of Fas ligand to T cell-mediated hepatic injury in mice. Gastroenterology 1997; 113: 1315–1322.
Kato J, Okamoto T, Motoyama H, Uchiyama R, Kirchhofer D, Van Rooijen N et al. Interferon-gamma-mediated tissue factor expression contributes to T-cell-mediated hepatitis through induction of hypercoagulation in mice. Hepatology 2013; 57: 362–372.
Ni HM, Chen X, Ding WX, Schuchmann M, Yin XM . Differential roles of JNK in ConA/GalN and ConA-induced liver injury in mice. Am J Pathol 2008; 173: 962–972.
Kunstle G, Hentze H, Germann PG, Tiegs G, Meergans T, Wendel A . Concanavalin A hepatotoxicity in mice: tumor necrosis factor-mediated organ failure independent of caspase-3-like protease activation. Hepatology 1999; 30: 1241–1251.
Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 2012; 19: 2003–2014.
Arshad MI, Piquet-Pellorce C, Filliol A, L'Helgoualc'h A, Lucas-Clerc C, Jouan-Lanhouet S et al. The chemical inhibitors of cellular death, PJ34 and Necrostatin-1, down-regulate IL-33 expression in liver. J Mol Med 2015; 93: 867–878.
Zhou Y, Dai W, Lin C, Wang F, He L, Shen M et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm 2013; 2013: 706156.
Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 2012; 3: e437.
Weinlich R, Oberst A, Dillon CP, Janke LJ, Milasta S, Lukens JR et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep 2013; 5: 340–348.
Suda J, Dara L, Yang L, Aghajan M, Song Y, Kaplowitz N et al. Knockdown of RIPK1 markedly exacerbates M 1 urine immune-mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of NF-kB. J Immunol 2016; 7: 1600690.
Saito K, Meyer K, Warner R, Basu A, Ray RB, Ray R . Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J Virol 2006; 80: 4372–4379.
Prikhod'ko EA, Prikhod'ko GG, Siegel RM, Thompson P, Major ME, Cohen JI . The NS3 protein of hepatitis C virus induces caspase-8-mediated apoptosis independent of its protease or helicase activities. Virology 2004; 329: 53–67.
Taniguchi H, Kato N, Otsuka M, Goto T, Yoshida H, Shiratori Y et al. Hepatitis C virus core protein upregulates transforming growth factor-beta 1 transcription. J Med Virol 2004; 72: 52–59.
Lim EJ, El Khobar K, Chin R, Earnest-Silveira L, Angus PW, Bock CT et al. Hepatitis C virus-induced hepatocyte cell death and protection by inhibition of apoptosis. J Gen Virol 2014; 95: 2204–2215.
Michelotti GA, Machado MV, Diehl AM . NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013; 10: 656–665.
Hirsova P, Gores GJ . Reply. Cell Mol Gastroenterol Hepatol 1: 265–266.
Hebbard L, George J . Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2011; 8: 35–44.
Brandon-Warner E, Schrum LW, Schmidt CM, McKillop IH . Rodent models of alcoholic liver disease: of mice and men. Alcohol 2012; 46: 715–725.
Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE . Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013; 57: 1773–1783.
Wang S, Ni HM, Dorko K, Kumer SC, Schmitt TM, Nawabi A et al. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016; 7: 17681–17698.
Bertola A, Mathews S, Ki SH, Wang H, Gao B . Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013; 8: 627–637.
Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A et al. Receptor interacting protein 3 protects mice from high fat diet-induced liver injury. Hepatology 2016; 64: 1518–1533.
Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci 2015; 129: 721–739.
Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 2014; 6: 1062–1074.
Pierce RH, Campbell JS, Stephenson AB, Franklin CC, Chaisson M, Poot M et al. Disruption of redox homeostasis in tumor necrosis factor-induced apoptosis in a murine hepatocyte cell line. Am J Pathol 2000; 157: 221–236.
Jones BE, Lo CR, Liu H, Srinivasan A, Streetz K, Valentino KL et al. Hepatocytes sensitized to tumor necrosis factor-alpha cytotoxicity undergo apoptosis through caspase-dependent and caspase-independent pathways. J Biol Chem 2000; 275: 705–712.
Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A . Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol 1994; 153: 1778–1788.
Hirsova P, Gores GJ . Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol 2015; 1: 17–27.
Kohli R, Feldstein AE . NASH animal models: are we there yet? J Hepatol 2011; 55: 941–943.
Chalasani NP, Hayashi PH, Bonkovsky HL, Navarro VJ, Lee WM, Fontana RJ et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol 2014; 109: 950–966; quiz 67.
Dara L, Liu ZX, Kaplowitz N . Mechanisms of adaptation and progression in idiosyncratic drug induced liver injury, clinical implications. Liver Int 2015; 36: 158–165.
Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014; 513: 90–94.
Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 2014; 513: 95–99.
Estornes Y, Aguileta MA, Dubuisson C, De Keyser J, Goossens V, Kersse K et al. RIPK1 promotes death receptor-independent caspase-8-mediated apoptosis under unresolved ER stress conditions. Cell Death Dis 2014; 5: e1555.
Schneider AT, Gautheron J, Tacke F, Vucur M, Luedde T . Receptor Interacting Protein Kinase-1 (RIPK1) in hepatocytes does not mediate murine acetaminophen toxicity. Hepatology 2016; 64: 306–308.
Acknowledgements
This work was supported by NIH Grant K08DK109141 (LD), R01DK067215 (NK), P30DK048522 (NK), and U01AA021857 (Z-XL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Silke
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dara, L., Liu, ZX. & Kaplowitz, N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discovery 2, 16089 (2016). https://doi.org/10.1038/cddiscovery.2016.89
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/cddiscovery.2016.89
This article is cited by
-
Mediators of necroptosis: from cell death to metabolic regulation
EMBO Molecular Medicine (2024)
-
A RIPK3-independent role of MLKL in suppressing parthanatos promotes immune evasion in hepatocellular carcinoma
Cell Discovery (2023)
-
RIPK3 promoter hypermethylation in hepatocytes protects from bile acid-induced inflammation and necroptosis
Cell Death & Disease (2023)
-
Prolonged hypoxia alleviates prolyl hydroxylation-mediated suppression of RIPK1 to promote necroptosis and inflammation
Nature Cell Biology (2023)
-
RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages
Cell Death & Differentiation (2021)
