
Abstract
We present preliminary data on mitochondrial DNA diversity within and among populations of the ants Lasius niger and Lasius platythorax in Poland. Phylogenetic analysis based on the mitochondrial DNA markers: cytochrome c oxidase subunit I (cox1) and 16S ribosomal RNA (16S rRNA) confirms the species status of L. niger and L. platythorax. Intraspecific variability is low in both species, which might be a result of severe bottlenecks and rapid postglacial expansion into Central Europe.
Similar content being viewed by others


Introduction
The sibling species Lasius niger and Lasius platythorax are widely distributed throughout the Palearctic ecozone. The latter species was described only as late as 1991 by Seifert. Despite their overall morphological similarity, the two species can be separated by discriminant analysis with various morphometric characters (Seifert, 2007). Lasius niger specializes in open habitats and nests mainly in soil, while L. platythorax is more common in woodland habitats and nests often in rotten wood. The two species are well separated with regard to ecological parameters (Seifert, 2007).
There have been few molecular studies aiming to test phylogenetic relationships among Formicidae taxa including Lasius ants (Hasegawa, 1998; Hasegawa et al., 2002; Janda et al., 2004; Steiner et al., 2004; Maruyama et al., 2008). In these studies, only single sequences of individuals of L. niger species (mainly) and/or L. platythorax (rarely) were examined. Genetic data also were used to reconstruct the mating frequency of colony queens and paternity variation in the ant Lasius niger (Boomsma and Van der Have, 1998; Fjerdingstad et al., 2003). However, there is a lack of data on geographic distribution of genetic diversity in L. platythorax and these concerning L. niger are scarce. The phylogeography of ants has only recently received attention (Goropashnaya et al., 2004; Pusch et al., 2006).
Here, we present preliminary data on the mitochondrial DNA diversity within and among populations of the ants L. niger and L. platythorax in Poland. Using two mtDNA markers: cytochrome c oxidase subunit I (cox1) and 16S ribosomal RNA (16S rRNA), we aimed at refining genetic relationships of L. niger and L. platythorax.
Materials and methods
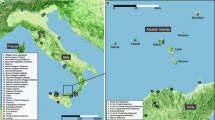
The ant samples (workers) were collected from nests situated in north-east, Central and Southern Poland (Table 1) and individual specimens were classified on the basis of the description given by Seifert (1992) and Radchenko et al. (1999). Each locality was assumed to comprise a population. Individuals from three L. niger populations and two L. platythorax populations distant by at least 300 km were analyzed. Total DNA was extracted from the frozen specimens with Genomic Mini kit (A&A Biotechnology). The cox1 and 16S rRNA gene fragments were amplified using HCO2198/LCO1490 (Folmer et al., 1994), and 16Sbr/16Sar-Dr (Palumbi and Benzie, 1991) primer pairs, respectively. The annealing conditions were set to 49°C for 1 min (cox1) and 52°C for 1 min (16S rRNA). PCR products were sequenced directly from both directions, using the same primers as at the amplification stage.
All newly obtained sequences were deposited in GenBank (accession nos: GQ503244–GQ503250). Sequence alignment was achieved with BioEdit (Hall, 1999). For the phylogenetic analyses, 680 bp of cox1 and 401 bp of 16S rRNA fragments were used. To infer phylogenetic relationships, neighbour-joining algorithm (NJ), maximum parsimony analysis (MP) with MEGA 4.0 (Tamura et al., 2007) and maximum likelihood method (ML) with TREE-PUZZLE 5.1 (Schmidt et al., 2002) were performed. Tamura-Nei distance (DTrN; Tamura and Nei, 1993) was used for the NJ trees. Statistical confidence in nodes was calculated by bootstrap resampling with 1,000 replicates (Felsenstein, 1985) or quartet puzzling steps for TREE-PUZZLE.
Based on programs ModelTest 3.1 (Posada and Crandall, 1998) and PAUP (Swofford, 2002) the substitution model Tamura-Nei (TrN), with equal rates for all sites in both dataset (cox1 and 16S rRNA), was used. Data from both regions were analyzed separately and then combined and analysed together (Fig. 1). Based on the phylogenetic studies for ants (Steiner et al., 2004; Maruyama et al., 2008) Lasius emarginatus sequences were used as an outgroup (accession nos: AB371057, AY225868).

Neighbour-joining (NJ) phylogenetic tree of combined data of region cox1 + 16S rRNA calculated with TrN algorithm. Number after species name indicates localities for particular haplotype/population. Bootstrap values >65 were given at nodes for NJ and MP (first and second values) and quartet puzzling steps for ML phylogenetics methods (third value)
The analyses were also complemented by adding previously published sequences of the gene regions targeted, retrieved from GenBank: cox1; accession nos.: AB007981 (Hasegawa, 1998; Hasegawa et al., 2002), AY225866–AY225867 (Steiner et al., 2004), AB371019-AB371020 (Maruyama et al., 2008) and 16S rRNA; accession nos.: AB371065-AB371066 (Maruyama et al., 2008). However, for the cox I sequence alignments the 379 bp overlap with sequences reported by Steiner et al. (2004) and the 283 bp overlap with sequences reported by Maruyama et al. (2008) were possible. For 16S rRNA data, the overlap with sequences deposited in GenBank was complete (401 bp).
Results
In case of cox1 gene region, analyses were based on 14 variable sites (12 informative sites) for only third codon position. All mutational changes were base pair substitutions, with about 71.4% of Tis. No mutation led to a change at the amino acid level. Within L. niger samples, two different haplotypes were detected with a single base change separating L. niger 77 colony (accession no: GQ503248) from the rest of colonies (Fig. 1). A similar situation was observed also in the case of L. platythorax; L. platythorax 102 and 103 colonies (accession no: GQ5032549) show a single nucleotide substitution (Fig. 1). Table 2 reports the pairwise distance DTrN values. The sequences were shortened (different pairs of primers were used in the L. emarginatus studies); however, the distance values remained unchanged.
In case of 16S rRNA region, analyses were based on eight variable sites (seven informative sites). All mutational changes were base pair substitutions, with about 75% of Tis. Within L. niger colonies, two different haplotypes were found with a single base change separating L. niger 68 colony (accession no: GQ503245) from the rest of L. niger colonies (Fig. 1). In L. platythorax samples, only one representative haplotype was observed. Table 3 reports the pairwise distance DTrN values.
As both mtDNA regions analysed gave the same result, the data were pooled and one combined phylogenetic tree was constructed (Fig. 1). The congruence of the tree topologies generated by different phylogenetic methods suggests that this is a reliable estimate of true relationships.
Discussion
The sequences of the cox I and 16S rRNA regions for all the specimens studied were found to fall into two monophyletic groups. The range of sequence divergences (Tables 2, 3) between L. niger and L. platythorax suggests that both species have been well separated though the time of interspecific diversification appears to be a rather recent. If we applied (after Pusch et al., 2006) the honey bee rate of mtDNA divergence, 2% per million years (Arias and Sheppard, 1996), these species would have diverged about 1 Mya.
The recent morphological and genetic data revealed the surprising commonness of hybridization in ants (Feldhaar et al., 2008; Seifert, 2009), also among members of the genus Lasius (Pearson, 1983; Seifert, 1999; Umphrey, 2006). It seems plausible that in case of recently diverged species, as L. niger and L. platythorax, the reproductive barriers cannot completely prevent the possibility of interspecific hybridization. There are reports that both species L. niger and L. platythorax may hybridize with other ant species, i.e., L. niger with L. alienus (Pearson, 1983) or L. psammophilus (Seifert, 1999), and L. platythorax with L. emarginatus (Seifert, 1999). There are even some observations of morphologically intermediate forms between L. niger and L. platythorax inhabiting the moss-covered rock cracks in Finland (W. Czechowski, pers. comm.). However, there is no scientific evidence of hybridization between both Lasius species.
With respect to geography, the hybridization may occur sporadically between broadly sympatric species or be limited to particular contact zone (Avise, 2004). In case of L. niger and L. platythorax, both species occur in close geographic proximity and also our observations confirm their sympatry; the colonies of both species can be situated even less than 50 m apart. On the other side, ant species which maintained interspecific contact during their natural history have built up more effective mechanisms of prezygotic isolation than species that rarely or never experienced such contact (Seifert, 1999). In consequence, the observed frequency of hybridization is lower in the first group of species (Seifert, 1999). The chances for interspecific hybridization may also depend on the time limitation for mating activities. L. niger and L. platythorax do not differ in the timing of their sexual activity, resulting in the possibility of co-occurrence of sexuals of both species. Moreover, in the north-eastern part of Poland we observed that even the daily hours of their nuptial flights may overlap. Both sibling species are monogynous and monoandrous, what possibly leaves less opportunities for interspecific hybridization. However, in southern Germany, Temnothorax hybrid workers were frequently found among workers of pure-species T. nylanderi or T. crassispinus, despite obligate monogyny and monandry in both sibling species (Pusch et al., 2006). Although there are no detailed studies, the hybridization between L. niger and L. platythorax seems also probable and only detailed research based on more samples and a wider study area could shed more light on this topic.
Along with marked interspecies divergence between L. niger and L. platythorax, low intraspecific variation across the examined range was observed. In our analyses of combined data (cox I + 16S rRNA), 57% of L. niger haplotypes were identical and 60% of L. platythorax. In comparison, two other sibling ant species widespread in Europe: T. crassispinus and T. nylanderi, showed similar genetic divergence values in cox I haplotypes (2.4%) but no more than 40% haplotypes of both species were identical (Pusch et al., 2006). Moreover, when we complemented our analyses by adding the cox I and 16S rRNA sequences obtained from specimens of L. niger and L. platythorax from Austria (Steiner et al., 2004; Maruyama et al., 2008), we found that despite the long geographical distance, the most common Polish haplotypes of both species appeared to be identical to previously mentioned haplotypes deposited in GenBank.
In one case, the cox I haplotype of L. niger (Hasegawa, 1998; Hasegawa et al., 2002), we noted the 3% sequence divergence (for 470 bp sequence alignment) between the haplotype mentioned and the cluster of L. niger and L. platythorax Polish haplotypes. If we assumed that this haplotype represents correctly classified species, such a level of L. niger intraspecific variation could be the result of local differentiation processes, often observed for geographically widely distributed species (e.g., Ross and Shoemaker, 2005; Burns et al., 2008). However, the place of origin of the L. niger sample concerned appears to be unclear. The sample comes either from Spain (Hasegawa (1998) or from Germany (Hasegawa et al., 2002). This uncertainty makes inferring hardly possible.
Generally, geographically restricted species are characterized by lower genetic diversity, than more widespread species. However, in the present study we observed widely distributed common haplotype and few unique haplotypes that resemble typical star-like phylogeny. This phylogeny pattern and low diversity in both species strongly suggest a recent bottleneck event followed by, most probably postglacial, population expansion (e.g. Goropashnaya et al., 2004). Our results showing low intraspecific variation are also concordant with the preliminary allozyme analyses of four gene loci (Pgm, Idh, Mdh, Pgi) from Polish samples of L. niger and L. platythorax (A. Wysocka, unpubl. data). However, there is not enough data to draw inferences about the level of gene flow and introgression between the populations studied.
References
Arias M.C. and Sheppard W.S. 1996. Molecular phylogeny of honey bee subspecies (Apis mellifera L.) inferred from mitochondrial DNA sequence. Mol Phylogenet Evol. 5: 557–566
Avise J.C. 2004. Molecular Markers, Natural History and Evolution. Sinauer Associates, Sunderland, MA. 684 pp
Boomsma J.J. and Van der Have T.M. 1998. Queen mating and paternity variation in the ant Lasius niger. Mol. Ecol. 7: 1709–1718
Burns J.M., Janzen D.H., Hajibabaei M., Hallwachs W. and Hebert P.D.N. 2008. DNA barcodes and cryptic species of skipper butterflies in the genus Perichares in Area de Conservación Guanacaste, Costa Rica. Proc. Natl. Acad. Sci. USA 17: 6350–6355
Feldhaar H., Foitzik S. and Heinze J. 2008. Life-long commitment to the wrong partner: hybridization in ants. Phil. Trans. R. Soc. B 363: 2891–2899
Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791
Folmer O., Black M., Moeri W., Lutz R. and Vrijenhoek R. 1994. DNA primers for amplifications of mitochondrial cytochrome oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3: 129–199
Fjerdingstad E.J., Gertsch P.J. and Keller L. 2003. The relationship between multiple mating by queens, within-colony genetic variability and fitness in the ant Lasius niger. J. Evol. Biol. 16: 844–853
Goropashnaya A.V., Fedorov V.B. and Pamilo P. 2004. Recent speciation in the Formica rufa group ants (Hymenoptera, Formicidae): inference from mitochondrial DNA phylogeny. Mol. Phylogenet. Evol. 32: 198–206
Hall T.A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis programm for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41: 95–98
Hasegawa E. 1998. Phylogeny and host-parasite relationships in social parasitism in Lasius ants. Entomol. Science 1: 133–135
Hasegawa E., Tinaut A. and Ruano F. 2002. Molecular phylogeny of two slave-making ants: Rossomyrmex and Polyergus (Hymenoptera: Formicidae). Ann. Zool. Fennici 39: 267–271
Janda M., Folková D. and Zrzavý J. 2004. Phylogeny of Lasius ants based on mitochondrial DNA and morphology, and the evolution of social parasitism in the Lasiini (Hymenoptera: Formicidae). Mol. Phylogenet. Evol. 33: 595–614
Maruyama M., Steiner F.M., Stauffer C., Stauffer C., Akino T., Crozier R.H. and Schlick-Steiner B.C. 2008. A DNA and morphology based phylogenetic framework of the ant genus Lasius with hypotheses for the evolution of social parasitism and fungiculture. BMC Evol. Biol. 8: 237–251
Palumbi S.R. and Benzie J. 1991. Large mitochondrial DNA differences among morphologically similar Penaeid shrimp. Mol. Mar. Biol. Biotechnol. 1: 27–34
Pearson B. 1983. Hybridization between the ant species Lasius niger and Lasius alienus - the genetic evidence. Insect. Soc. 30: 402–411
Posada D. and Crandall K.A. 1998. Modeltest: testing the model on DNA substitution. Bioinformatics 14: 817–818
Pusch K., Seifert B., Foitzik S. and Heinze J. 2006. Distribution and genetic divergence of two parapatric sibling ant species in Central Europe. Biol. J. Linn. Soc. 88: 223–234
Radchenko A., Czechowska W., Czechowski W. and Siedlar E. 1999. Lasius niger (L.) and Lasius platythorax Seifert (Hymenoptera, Formicidae) - a revolution in Polish myrmecological faunistics and zoocenology? Fragm. Faun. 42: 103–113
Ross K.G. and Shoemaker D.D. 2005. Species delimitation in native South American fire ants. Mol. Ecol. 14: 3419–3438
Schmidt H.A., Strimmer K., Vingron M. and von Haeseler A. 2002. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18: 502–504
Seifert B. 1992. A taxonomic revision of the Palaearctic members of the ant subgenus Lasius s. str. (Hymenoptera: Formicidae). Abh. Ber. Naturkundemus. Görlitz 66: 1–67
Seifert B. 1999. Interspecific hybridisations in natural populations of ants by example of a regional fauna (Hymenoptera, Formicidae). Insect. Soc. 46: 45–52
Seifert B. 2007. Die Ameisen Mittel-und Nordeuropeas. Lutra, Klitten. 368 pp
Seifert B. 2009. Cryptic species in ants (Hymenoptera: Formicidae) revisited: we need a change in the alpha-taxonomic approach. Myrmecol. News 12: 149–166
Steiner F.M., Schlick-Steiner B.C., Schödl S., Espadaler X., Seifert B., Christian E. and Stauffer C. 2004. Phylogeny and bionomics of Lasius austriacus (Hymenoptera, Formicidae). Insect. Soc. 51: 24–29
Swofford D.L. 2002. PAUP* Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b10 - Sinauer Associates, Sunderland, Massachusetts
Tamura K., Dudley J., Nei M. and Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Biol. Evol. 24: 1596–1599
Tamura K. and Nei M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10: 512–526
Umphrey G.J. 2006. Sperm parasitism in ants: selection for interspecific mating and hybridization. Ecology 87: 2148–2159
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wysocka, A., Krzysztofiak, L., Krzysztofiak, A. et al. Low genetic diversity in Polish populations of sibling ant species: Lasius niger (L.) and Lasius platythorax Seifert (Hymenoptera, Formicidae). Insect. Soc. 58, 191–195 (2011). https://doi.org/10.1007/s00040-010-0135-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00040-010-0135-9