
Abstract
The internal energies and entropies of 21 well-known minerals were calculated using the density functional theory (DFT), viz. kyanite, sillimanite, andalusite, albite, microcline, forsterite, fayalite, diopside, jadeite, hedenbergite, pyrope, grossular, talc, pyrophyllite, phlogopite, annite, muscovite, brucite, portlandite, tremolite, and CaTiO3–perovskite. These thermodynamic quantities were then transformed into standard enthalpies of formation from the elements and standard entropies enabling a direct comparison with tabulated values. The deviations from reference enthalpy and entropy values are in the order of several kJ/mol and several J/mol/K, respectively, from which the former is more relevant. In the case of phase transitions, the DFT-computed thermodynamic data of involved phases turned out to be accurate and using them in phase diagram calculations yields reasonable results. This is shown for the Al2SiO5 polymorphs. The DFT-based phase boundaries are comparable to those derived from internally consistent thermodynamic data sets. They even suggest an improvement, because they agree with petrological observations concerning the coexistence of kyanite + quartz + corundum in high-grade metamorphic rocks, which are not reproduced correctly using internally consistent data sets. The DFT-derived thermodynamic data are also accurate enough for computing the P–T positions of reactions that are characterized by relatively large reaction enthalpies (> 100 kJ/mol), i.e., dehydration reactions. For reactions with small reaction enthalpies (a few kJ/mol), the DFT errors are too large. They, however, are still far better than enthalpy and entropy values obtained from estimation methods.
Similar content being viewed by others


Introduction
Investigating the standard enthalpy of formation from the elements (ΔfH298.15) and the standard entropy (S298.15) of mineral end members of geological and cosmochemical relevance is a prerequisite for reliable phase diagram calculations and still needed in many aspects: first, because such standard data of some mineral end members are derived from a limited number of experiments and largely missing for chemically more complex systems (e.g., Ti-containing end members of many solid solutions such as pyroxenes, micas, amphiboles) and, second, because there are important mineral solid solutions whose thermodynamic description needs data from end members which do not exist physically at all (e.g., Al-rich biotite end members). The experimental data basis is also poor for rare parageneses (e.g., minerals in calcium–aluminium-rich inclusions of primitive meteorites). This has the consequence that thermodynamic calculations including such end members suffer from missing accuracy.
Nowadays, the generation of such end member thermodynamic data is possible in a relatively short time by using the density functional theory (DFT). These data can then be integrated into existing thermodynamic data sets making them available to a broad range of applications.
The following question, however, arise: Are the DFT results accurate enough?
The uncertainties in ΔfH298.15 given in internally consistent thermodynamic data sets are in the order of a few kJ/mol for well-known phases. Because ΔfH298.15 data of such mineral end members are based on a large number of experimental data, it can be assumed that their accuracy is of the same order. The uncertainties of reaction enthalpies derived from solution calorimetry is around a few kJ/mol (e.g., Majzlan et al. 2017), and those of standard entropy values derived from relaxation calorimetry is in the order of a few J/mol/K and does not exceed 1% (Dachs and Benisek 2011). The question of the required accuracy of DFT-calculated standard data needed for reliable phase diagram calculations, is, therefore, easy to answer. It should not be much larger than the uncertainties of the two calorimetric methods mentioned, allowing incorporating them in the development of internally consistent databases. The quality of DFT-calculated thermodynamic data should therefore be tested by comparison with those from well-known mineral end members with their high accuracy.
The comparison of DFT-calculated energies to enthalpies listed in thermodynamic data sets is, however, directly not possible, first, because their zero levels are different. In thermodynamic data sets, the enthalpies of the elements are set to zero, whereas in DFT calculations, the atom cores separated from the valence electrons have zero potential. The most obvious way for solving this problem is to investigate the DFT energies of the minerals in question as well as of their constituting elements and to calculate then the formation energies from the elements. Since the elements and most mineral end members belong to quite different classes of materials, this approach leads to large errors (Benisek and Dachs 2017). A more appropriate approach is to calculate the energy of the phases under consideration, as well as of the binary oxides they are made of (Hautier et al. 2012). This enables the determination of the formation energy of a phase from its constituting oxides. Adding ΔfH values of the oxides, which are tabulated with high accuracy in the Janaf-tables (Chase 1998), the ΔfH value of a phase from the elements can be determined. A second problem for the comparison of tabulated and DFT-based ΔfH values is the fact that energies calculated by the DFT method are internal energies at 0 K, which need to be transformed into enthalpies at 298.15 K.
The standard entropy of a phase can also be calculated by the DFT method by investigating the lattice dynamics, which results in heat capacity data. This heat capacity, however, is that at constant volume (CV) and the entropy derived from it is not directly comparable to S298.15 of thermodynamic data sets, which is based on the heat capacity at constant pressure (CP). Principally, CP could be computed by the DFT method by using the quasiharmonic approximation (Born and Huang 1956), which is, however, a time-consuming method. If this method must be avoided, CV has to be transformed into CP somehow different to make the entropies comparable.
Since the use of the quasiharmonic approximation is found to be too time-consuming for a broad study on the accuracy of DFT-calculated enthalpies and entropies, we offer in this publication an efficient method to transform these quantities, enabling a direct comparison between DFT-calculated and tabulated properties on a large number of minerals. Such a comparison on many well-known mineral end members is needed to answer the question, if DFT-calculated thermodynamic properties are able to complement calorimetric experiments. The errors generated by the transformation method should be much smaller than those generated by the DFT method itself to make a reliable comparison possible.
In this study, we present DFT-based ΔfH298.15 and S298.15 values of 21 well-known mineral end members. These data were applied to phase equilibrium calculations to assess the associated uncertainties and to discuss relevant petrological problems.
Experimental methods
Computational methods
Quantum–mechanical calculations were based on the DFT plane wave pseudopotential approach implemented in the CASTEP code (Clark et al. 2005) included in the Materials Studio software from Accelrys®. The calculations used the local density approximation (LDA) for the exchange–correlation functional (Ceperley and Alder 1980) and norm-conserving pseudopotentials to describe the core–valence interactions. For the k-point sampling of the investigated unit cells, a Monkhorst–Pack grid (spacing of 0.02/Å) was used (Monkhorst and Pack 1976) and convergence was tested by performing calculations using a denser k-point grid. The structural relaxation was calculated applying the BFGS algorithm, where the threshold for the force on the atom was 0.01 eV/Å. The calculations on Fe-containing minerals used an ultrasoft pseudopotential and the LDA + U approach (with U = 4.0 eV applied to Fe d orbitals). In addition to the LDA calculations, the gradient-corrected functional (GGA-PBE) from Perdew et al. (1996), its revised form for solids (GGA-PBESOL, Perdew et al. 2008), and hybrid functionals such as B3LYP (Becke 1993) and PBE0 (Adamo 1999) were used for comparison reasons in some cases. Using the hybrid functionals, the single point energies were calculated on a LDA-geometry optimized cell.
The lattice dynamical calculations were performed for the relaxed structures within the linear response approximation implemented in CASTEP using the interpolation approach and a wider k-point grid (spacing of 0.05/Å) compared to the energy calculations. The lattice dynamical calculations on Fe-containing minerals were based on the finite displacement approach, which calculated the forces on perturbed configurations in a super cell with positive and negative displacements.
Transformation of DFT-calculated quantities to those tabulated in thermodynamic data sets
Heat capacity
The difference between the heat capacity at constant volume (CV) and that at constant pressure (CP) is given by the following equation (e.g., Cemic 2005):
where V denotes the molar volume, α the thermal expansion coefficient, and KT the isothermal bulk modulus. To make the transformation from CV to CP, the values for V, α, and KT are assumed to be similar within a given mineral group (e.g., pyroxenes, micas) so that the relative difference between CV and CP
can be averaged for a particular mineral group. This enables that DFT-calculated CV of a phase belonging to this family can be transformed to CP.
Enthalpy and entropy
The DFT method was used to compute the internal reaction energy at 0 K (ΔRU0K) of the formation of a mineral phase from its oxides. Based on the generalized reaction,
ΔRU0K is given by:
where U0KAO, \({U^{0{\text{K}}}}_{{{\text{B}}{{\text{O}}_{\text{2}}}}}\), and \({U^{0{\text{K}}}}_{{{{\text{A}}_{\text{2}}}{\text{B}}{{\text{O}}_{\text{4}}}}}\) are the internal energies of the reactants at 0 K.
As a next step, it was assumed that ΔRU0K of reaction (3) was equal to the reaction enthalpy at 0 K (ΔRH0K), i.e.,
Since ΔRU0K is the reaction energy of solids at 0 K, this simplification does not lead to relevant errors because the volume term (PΔRV) relating internal energy and enthalpy is very small and can be neglected (Hautier et al. 2012). The enthalpy of formation of A2BO4 from the elements at 0 K \(\left( {{\Delta _{\text{f}}}{H^{0{\text{K}}}}_{{{{\text{A}}_{\text{2}}}{\text{B}}{{\text{O}}_{\text{4}}}}}} \right)\) was then calculated using accurate ΔfH0K values of the constituting oxides taken from the Janaf-tables (Chase 1998) according to:
The so-derived values contain only simplifications inherent in the DFT calculations. Using the above described approach to convert CV to CP (Sect. “Heat capacity”), the standard enthalpy was then computed via
A sketch of the complete calculation procedure is shown in Fig. 1 for forsterite as an example. The entropy was calculated by integrating the calculated CP/T values over the temperature range from 0 to 298.15 K, i.e.,

Sketch of the calculation procedure for forsterite as an example. The following reaction was investigated: 2MgO + SiO2 = Mg2SiO4. Its reaction energy at 0 K (ΔRU0K) was calculated by the DFT method and it is assumed that the reaction enthalpy at 0 K (ΔRH0K) is identical. Adding the formation enthalpies of the oxides at 0 K (ΔfH0Kox) to this ΔRH0K value results in the formation enthalpy of forsterite from the elements at 0 K (ΔfH0Kfo). The heat content from 0 to 298.15 K (= ∫CPfo dT) is than added to this value yielding finally the standard enthalpy of formation of forsterite from the elements (ΔfH298.15fo)
Fe-bearing minerals
For Fe-containing minerals, another transformation method was used to bypass problems caused by the magnetic phase transitions appearing in these systems, since the magnetic entropy cannot be determined directly with DFT (Hickel et al. 2011). The DFT-calculated reaction energies and entropies from the oxides (Eq. 3) at 0 K were transformed to 298.15 K using the calculated reaction heat capacities (ΔRCV) of Eq. (3). As shown later, the integrals of ΔRCV and ΔRCV/T over temperature have similar values when compared to those of measured ΔRCP and ΔRCP/T. This is because the effect of magnetic transformations on the heat capacity are present in both, the right and the left hand side of Eq. (3), thus partly eliminating their magnetic contribution. The same is true with the difference between heat capacity at constant volume and that at constant pressure. The reaction enthalpies and entropies at 298.15 K of mineral end members from their oxides were calculated according to
Finally, ΔfH298.15 and S298.15 of Fe-containing minerals were then calculated using the oxide data at 298.15 K from the Janaf-tables (Chase 1998).
OH-containing minerals
Several minerals containing OH groups were investigated. Consequently, reaction (3) has H2O on its left hand side. A directly calculated internal energy of H2O at 0 K using the DFT method showed, however, a mean systematic error of 38 kJ/mol. The DFT-based internal energy of H2O at 0 K was, therefore, shifted by this value improving the ΔfH298.15 values of the investigated OH-containing mineral end members.
Investigated mineral end members and their structural models
The investigated minerals are listed with formulae and abbreviations in Table 1. The atomic distributions are fully ordered for most of the investigated minerals and there is no configurational contribution to the entropy for these cases. However, biotite end members and muscovite are characterized by a disordered distribution of Al and Si on tetrahedral sites. For these minerals, we investigated a larger number of cells having all possible configurations except the ones, where Al–O–Al bonds are present. This Al-avoidance treatment is in accordance with numerous studies (e.g., Circone et al. 1991; Palin et al. 2001; Vinograd and Putnis 1998), which all found that adjacent Al tetrahedra in micas are virtually absent. From the DFT results of all investigated cells, the mean values of CP and ΔfH298.15 were calculated, which assumes that all investigated configurations are present and equally probable. The impact of this assumption on the accuracy of ΔfH298.15 and S298.15 was estimated to be in the order of kJ/mol and J/mol/K, respectively. The configurational entropy was calculated with a Al-avoidance model (e.g., Holland and Powell 1998) and was added to the vibrational entropy calculated by the DFT method.
Results
Heat capacity
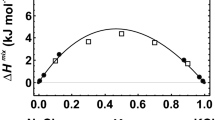
The relative difference between CV and CP (Eq. 2) is shown in Fig. 2 for the pyroxenes as an example. The CP data were taken from the literature, and CV was calculated by the DFT method showing a linear relationship of ΔCrel versus T (solid line in Fig. 2). The DFT-calculated CV of the pyroxenes can thus easily be transformed to CP. This procedure was performed for the other mineral groups as well. The heat capacities of all investigated minerals are compared to reference values in Table 2 showing that deviations are in the order of a few J/mol/K. The impact of the adopted CV−CP conversion on ΔfH298.15 and S298.15 is in the order of 0.1 kJ/mol and 1 J/mol/K, respectively, and is shown for ΔfH298.15 of low microcline in the next chapter. Using this procedure, DFT-calculated CV values of unknown members of the mineral groups studied can be transformed to the corresponding CP values.

Relative mean difference between heat capacity at constant pressure (CP) and that at constant volume (CV) plotted as a function of temperature for the pyroxenes diopside and jadeite. Error bars represent 1 sd
As already mentioned in Sect. “Fe-bearing minerals’’, Fe-containing minerals were calculated using a different routine. In Table 3, the integrals of the calculated ΔRCV and ΔRCV/T over temperature are compared to those of measured ΔRCP and ΔRCP/T, showing acceptable agreement. The impact of the simplifications adopted in this procedure on the accuracy of ΔfH298.15 and S298.15 were estimated to be in the order of 1 kJ/mol for the enthalpy and some J/mol/K for the entropy.
Enthalpy
The results of the DFT calculations are summarized and compared to reference values in Table 4. Almost all DFT ΔfH298.15 values are slightly more negative (by a few kJ/mol) than the reference values. The deviation is ~ 7 kJ/mol in average and does not exceed 20 kJ/mol for most minerals. One outlier is pyrope with a deviation of 55.83 kJ/mol. Possible reasons for that are unknown.
The errors generated by the transformation of CV to CP can be directly estimated by comparing DFT-calculated ΔRU0K values with measured ΔRH0K values. This comparison was undertaken for low microcline as an example and is listed in Table 5. ΔRU0K is less negative by 4.4 kJ/mol than the measured ΔRH0K value. On the other hand, the calculated standard enthalpy of this study is also less negative by 4.43 kJ/mol (Table 4) when compared to the tabulated value. This demonstrates that the DFT calculation itself produces a deviation of ~ 4.4 kJ/mol from the experimental standard enthalpy, and not the simplifications made by transforming CV to CP.
Entropy
The calculated S298.15 values are shown in Table 4 and do not deviate more than 4.25 J/mol/K from the reference values except for two outliers. One outlier is again pyrope and the other is talc with deviations of − 8.31 and 7.37 J/mol/K, respectively. The ΔfH298.15 and S298.15 deviations of pyrope are partly compensated, when the free Gibbs energy (G) is considered, but this is not the case for talc, because here the deviation of H is negative, but that of S is positive.
Application of the new DFT-calculated H, S, and C P data
The calculated ΔfH298.15, S298.15, and CP data were tested on various mineral reactions to clarify the impact of their deviations from reference values on phase diagram calculations, thereby taking the values for the molar volume, thermal expansion and bulk modulus from the thermodynamic data set of Holland and Powell (2011). For generating these values by the DFT method, see Sect. “Application of the DFT method to calculate enthalpies and entropies of mineral end members not listed in internally consistent data bases”.
One-component systems: phase transitions
Earlier investigations (Benisek and Dachs 2018; Dachs et al. 2018) indicated that the reaction enthalpies (ΔH) of phase transitions calculated applying DFT methods are accurate. This can be tested using the new data on the Al2SiO5 system. Although the new ΔfH298.15 values of andalusite, kyanite, and sillimanite show deviations of more than 10 kJ/mol from reference values, they all have similar deviations. As a consequence, the new ΔH values of the phase transitions
and
do not deviate by more than 1.8 kJ/mol from those of the internally consistent thermodynamic data set of Holland and Powell (2011). To show the impact of these remaining deviations, the new ΔfH298.15, S298.15, and CP data were used to calculate the Al2SiO5 phase relations and compare it with those derived from the data of Holland and Powell (2011) as shown in Fig. 3. The triple point, where all Al2SiO5 phases are in equilibrium, is shifted to a slightly higher temperature and lower pressure. The most obvious differences are seen in the slopes of the curves. Some effort was thus undertaken to improve the agreement by testing other functionals than LDA. Although other functionals may lead to better agreement in the absolute values, the delta values of the phase transitions could not be improved. This can be judged from the values listed in Table 6. Using the GGA-PBESOL and PBE0 functionals, the calculated formation energy of kyanite from its oxides shows better agreement with the reference value compared to the LDA functional. Considering the energy difference of the phase transition kyanite = sillimanite, the LDA functional is much better than the other ones. It has been reported that the GGA functional leads overall to an improved agreement with experiment compared to the LDA functional (e.g., Stampfl et al. 2001). Considering differences (ΔH, ΔCP, etc.); however, our experiences are that the LDA functional is slightly superior over the others (e.g., Benisek and Dachs 2012, 2018; Benisek et al. 2018, this study).

Phase diagram of the Al2SiO5 system using DFT-calculated standard enthalpy, entropy, and heat capacity values of this study compared to calculations with the Holland and Powell (2011) data set (HP11). The thick curve represents the breakdown reaction of kyanite to corundum + quartz (using the data set of Holland and Powell 2011). The shaded area shows the stability field of corundum + quartz, if DFT-based data are used for the Al2SiO5 phases. If internally consistent data sets are used instead, the coexistence of quartz + corundum is not possible
However, the differences seen in Fig. 3 have to be discussed from another point of view. Calculations based on internally consistent thermodynamic data bases disagree with petrological findings as described in Harlov and Milke (2002). Although there is petrological evidence that kyanite coexists with quartz and corundum, all available internally consistent data sets predict that this is not possible. At a pressure of 7 kbar for example, kyanite would react to quartz and corundum at a temperature of ~ 800 °C. At this temperature, however, kyanite is not stable with respect to sillimanite (Fig. 3). The kyanite = sillimanite curve must intersect the kyanite = quartz + corundum equilibrium to predict a coexistence of kyanite with quartz + corundum. When using internally consistent data bases, this, however, is not the case for the P–T range realized in crustal and upper mantle rocks. Extraordinarily, the DFT-based data set of the Al2SiO5 phases delivers a kyanite = sillimanite curve, which intersects the breakdown curve of kyanite to quartz and corundum (Fig. 3).
Multi component reactions
The calculations with the Al2SiO5 phases showed that the DFT method is able to yield accurate thermodynamic data for one-component systems. Now, we will show the impact of the absolute errors in ΔfH298.15 of an Al2SiO5 phase on a reaction boundary. The DFT-calculated ΔfH298.15 value of kyanite is more negative by 11.72 kJ/mol when compared to the reference value. Since the DFT-derived value is based on the formation from the oxides, this deviation must become effective when calculating the reaction:
At standard conditions, ΔH of this reaction is 6.98 kJ/mol (Holland and Powell 2011). The DFT-derived value (18.75 kJ/mol) differs more than 100% from this value. Computing the reaction curve with the DFT-based data shifts the breakdown of kyanite to temperatures as high as 2000 °C, far from being realistic. This reaction was thoroughly investigated by Harlov and Milke (2002) confirming the data from internally consistent data bases. It is, therefore, no doubt that the reaction curve of (14) lies in the P–T range as shown in Fig. 3 and that the DFT method gave reaction properties for this equilibrium far from being accurate enough. Obviously, the crystal chemistry of the oxides and the Al2SiO5 phases are too different so that the error inherent in the DFT method is too large, especially in cases where ΔH of the investigated reaction is small. In a next step, we now consider reactions that have a larger ΔH, so that the DFT errors have a smaller impact on the reaction curves, i.e., dehydration reactions.
Multi component reactions including H2O
To show the full impact of the DFT errors on the phase relations, the thermodynamic data of only one phase are replaced by DFT data for the reactions to follow. If the data of two or more phases would be replaced, than the errors become smaller in most cases because of their covariance, i.e., most of the enthalpic DFT data deviate negatively from the reference values and, thus, errors would partly be compensated. The results of the following reaction,
are shown in Fig. 4. One curve is calculated with ΔfH298.15, S298.15, and CP data for pyrophyllite as determined by the DFT method. This reaction has a ΔH of ca. 73 kJ/mol. The DFT-based ΔfH298.15 deviates by 15.98 kJ/mol from the reference value (Table 4) and this causes a shift of ca. 200 °C in the calculated P−T position (Fig. 4), a value which is still too large.

Dehydration reaction of pyrophyllite (pyp) using DFT-calculated standard enthalpy, entropy, and heat capacity values for pyp compared to calculations with the Holland and Powell (2011) data set (HP11) for all phases
The reactions,
were then investigated and the results are shown in Fig. 5. The ΔH’s of these reactions are larger than 100 kJ/mol and the DFT-based enthalpies deviate less than 10 kJ/mol from reference values (Table 4). As a consequence, the effect of the DFT errors on the reaction temperatures is small (Fig. 5).
From the errors of DFT-computed ΔfH298.15 and S298.15 that of the entropy has a smaller impact on the phase relations. This can be shown for sillimanite as an example. At a temperature of 1000 K, the error in S298.15 produces an error in the Gibbs free energy (G) of − 1.81 kJ/mol, whereas for that of the enthalpy an error in G of − 12.67 kJ/mol (see Table 4).

Dehydration reaction of micas: (1) annite + kyanite + 2 quartz = almandine + sanidine + H2O; (2) muscovite + quartz = sillimanite + sanidine + H2O; (3) phlogopite + sillimanite + 2 quartz = pyrope + sanidine + H2O. Calculations were done using DFT-derived standard enthalpy, entropy, and heat capacity values of annite, muscovite, and phlogopite. They are compared to calculations with the Holland and Powell (2011) data set (HP11) for all phases
Application of the DFT method to calculate enthalpies and entropies of mineral end members not listed in internally consistent data bases
To present DFT data for a less known end member, we chose ordered Al-buffonite (CaTi1/2Mg1/2AlSiO6), because it is an important pyroxene end member in calcium–aluminium-rich inclusions (CAIs) of primitive meteorites and their thermodynamic data were only investigated by a few studies (e.g., Sack and Ghiorso 1994, 2017). The DFT-based thermodynamic standard data (ΔfH298.15, S298.15) are listed in Table 7, where they are compared to those of Sack and Ghiorso (2017) showing excellent agreement supporting their work. If a value for the volume, V298.15, 1 bar, is required, it can be calculated from the DFT-based volume (V0K) in a similar manner to that described above for CP, i.e., by evaluating a relative mean difference between V298.15, 1 bar and V0K of the well-known pyroxene end members, diopside, jadeite, and hedenbergite:
The volume at 298.15 K of the unknown pyroxene is then given by
Values for thermal expansion and for the bulk modulus may be derived from averaging the values of the well-known phases of this particular mineral group. If, however, these values have to be determined accurately, i.e., for HP–HT phases, then the quasi harmonic approximation should be used instead (see, e.g., Belmonte 2017).
Comparison with estimation methods
The relative deviations of DFT-calculated enthalpies from reference values (Hcalc−Href)/Href are compared in Fig. 6 to those resulting from an enthalpy estimation method (van Hinsberg et al. 2005). Clearly, the DFT method yields by far more accurate results. The DFT-calculated ΔfH298.15 values deviate by not more than 1.3% from the reference values, whereas those derived from the estimation method show deviations of more than 10%. For almost all investigated phases, the DFT method is more accurate. A similar picture can be drawn for S298.15.

DFT versus estimation method of van Hinsburg et al. (2005). The relative deviation of the calculated standard enthalpy ((Hcalc−Href)/Href) from reference values (Holland and Powell 2011) are plotted for the minerals investigated in van Hinsburg et al. (2005). Squares represent the DFT data of this study, which are compared to the estimated values (filled circles). Open circles represent estimated values for minerals not investigated by the DFT method
Conclusions
DFT-based thermodynamic data are suitable to calculate reaction curves of phase transitions as well as of reactions, whose ΔH’s are large (> 100 kJ/mol). For other reactions, this method does not yield accurate enough thermodynamic data. DFT-calculated energies have uncertainties in the order of 10 kJ/mol, slightly larger than those of calorimetric methods. The DFT data are, however, far better than data generated by estimation methods. The optimal approach will be to use DFT-calculated thermodynamic data together with those from other sources (calorimetry, phase equilibrium experiments) to develop internally consistent databases.
References
Adamo C (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110:6158
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Belmonte D (2017) First principles thermodynamics of minerals at HP–HT conditions: MgO as a prototypical material. Minerals. https://doi.org/10.3390/min7100183
Benisek A, Dachs E (2012) A relationship to estimate the excess entropy of mixing: application in silicate solid solutions and binary alloys. J Alloys Compd 527:127–131
Benisek A, Dachs E (2017) Can density-functional calculations deliver accurate heats of formation for materials relevant in Geosciences? Meeting of the DMG, GeoBremen
Benisek A, Dachs E (2018) Thermodynamics of disordering in Au3Cu. J Alloys Compd 735:1344–1349
Benisek A, Dachs E, Kroll H (2014) Thermochemistry of the alkali feldspars: calorimetric study of the entropy relations in the low albite−low microcline series. Am Mineral 99:76–83
Benisek A, Dachs E, Grodzicki M (2018) Vibrational entropy of disorder in Cu3Au with different degrees of short-range order. Phys Chem Chem Phys 20:19441–19446. https://doi.org/10.1039/C8CP01656A
Born M, Huang K (1956) Dynamical theory of crystal lattices. International series of monographs on physics. Oxford at the Clarendon Press, Hong Kong
Cemic L (2005) Thermodynamics in mineral sciences. Springer, Berlin
Ceperley DM, Alder BJ (1980) Ground state of the electron gas by a stochastic method. Phys Rev Lett 45:566–569
Chase MW (1998) NIST-JANAF thermochemical tables. J Phys Chem Ref Data, Monograph No. 9, Washington, DC
Circone S, Navrotsky A, Kirkpatrick RJ (1991) Substitution of [6,4]Al in phlogopite: Mica characterization, unit-cell variation, 27Al and 29Si MAS-NMR spectroscopy, and Al-Si distribution in the tetrahedral sheet. Am Mineral 76:1485–1501
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MIJ, Refson K, Payne MC (2005) First principles methods using CASTEP. Z Kristallogr 220:567–570
Dachs E, Benisek A (2011) A sample-saving method for heat capacity measurements on powders using relaxation calorimetry. Cryogenics 51:460–464
Dachs E, Benisek A, Redhammer G (2018) P21/c-C2/c phase transition and mixing properties of the(Li, Na) FeGe2O6 solid solution: a calorimetric and thermodynamic study. J Chem Thermodyn 120:123–140
Dachs E, Benisek A (2015) Standard-state thermodynamic properties of annite, KFe3[(OH)2AlSi3O10], based on new calorimetric measurements. Eur J Mineral 27:603–616
Harlov DE, Milke R (2002) Stability of corundum + quartz relative to kyanite and sillimanite at high temperature and pressure. Am Mineral 87:424–432
Haselton HT, Robie RA, Hemingway BS (1987) Heat capacities of synthetic hedenbergite, ferrobustamite, and CaFeSi2O6 glass. Geochim Cosmochim Acta 51:2211–2217
Hautier G, Ong SP, Jain A, Moore CJ, Ceder G (2012) Accuracy of density functional theory in predicting formation energies of ternary oxides from binary oxides and its implication on phase stability. Phys Rev B 85:155208
Hickel T, Grabowski B, Körmann F, Neugebauer J (2011) Advancing DFT to finite temperatures: methods and applications in steel design. J Phys Condens Matter 24:053202
Holland TJB, Powell R (1998) An internally consistent thermodynamic dataset for phases of petrological interest. J Metamorph Geol 16:309–343
Holland TJB, Powell R (2011) An improved and extended internally consistent thermodynamic dataset for phases of petrological interest, involving a new equation of state for solids. J Metamorph Geol 29:333–383
Majzlan J, Grevel KD, Kiefer B, Nielsen UG, Grube E, Dachs E, Benisek A, White MA, Johnson MB (2017) Thermodynamic and crystal chemistry of rhomboclase, (H5O2)Fe(SO4)2·2H2O, and the phase (H3O)Fe(SO4)2 and implications for acid mine drainage. Am Mineral 102:643–654
Monkhorst HJ, Pack JD (1976) On special points for Brillouin zone integrations. Phys Rev B 13:5188
Palin EJ, Dove MT, Redfern SAT, Bosenick A, Sainz-Diaz CI, Warren MC (2001) Computational study of tetrahedral Al–Si ordering in muscovite. Phys Chem Minerals 28:534–544
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approxomation made simple. Phys Rev Lett 77:3865–3868
Perdew JP, Ruzsinszky A, Csonka GI, Vydrov OA, Scuseria GE, Constantin LA, Zhou X, Burke K (2008) Restoring the density-gradient expansion for exchange in solids and surfaces. Phys Rev Lett 100:136406
Robie RA, Hemingway BS (1984) Entropies of kyanite, andalusite, and sillimanite: additional constraints on the pressure and temperature of the Al2SiO5 triple point. Am Mineral 69:298–306
Robie RA, Finch CB, Hemingway BS (1982) Heat capacity and entropy of fayalite (Fe2SiO4) between 5.1 and 383 K: comparison of calorimetric and equilibrium values for the QFM buffer reaction. Am Mineralgist 67:463–469
Sack RO, Ghiorso MS (1994) Thermodynamics of multicomponent pyroxenes: III. Calibration of Fe2+(Mg)–1, TiAl2(MgSi2)–1, TiFe3 + 2(MgSi2)–1, AlFe3+(MgSi)–1, NaAl(CaMg)–1, Al2(MgSi)–1 and Ca(Mg)–1 exchange reactions between pyroxenes and silicate melts. Contrib Mineral Petrol 118:271–296
Sack RO, Ghiorso MS (2017) Ti3+—and Ti4+—rich fassaites at the birth of the solar system: thermodynamics and applications. Am J Sci 317:807–845
Stampfl C, Mannstadt W, Asahi R, Freeman AJ (2001) Electronic structure and physical properties of early transition metal mononitrides: Density-functional theory LDA, GGA, and screened-exchange LDA FLAPW calculations. Phys Rev B 63:155106
Van Hinsberg VJ, Vriend SP, Schumacher JC (2005) A new method to calculate end-member thermodynamic properties of minerals from their constituent polyhedral I: enthalpy, entropy and molar volume. J Methamorph Geol 23:165–179
Vinograd VL, Puntis A (1998) Calculation of the configurational entropy of Al, Si in layer silicates using the cluster variation method. Phys Chem Miner 26:136–148
Acknowledgements
Open access funding provided by Paris Lodron University of Salzburg. This work was supported by grants from the Austrian Science Fund (FWF), project number P 28724, which is gratefully acknowledged. We thank E. Forsthofer, P. Zinterhof, and colleagues for their professional work in implementing and maintaining the Materials Studio software at the Department of Computer Sciences, Salzburg University. We also acknowledge the careful reviews of two anonymous referees.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Mark S Ghiorso.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Benisek, A., Dachs, E. The accuracy of standard enthalpies and entropies for phases of petrological interest derived from density-functional calculations. Contrib Mineral Petrol 173, 90 (2018). https://doi.org/10.1007/s00410-018-1514-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-018-1514-x