
Abstract
Peanut (Arachis hypogaea), a major source of vegetable oil in many Asian countries, has become an integral part of human diet globally due to its high nutritional properties and option to consume in different forms. In order to meet the demand of vegetable oil, many peanut breeding programs of China have intensified their efforts in increasing oil content in newly bred varieties for reducing the import of edible oils in China. In this context, transcriptome sequencing data generated on 49 peanut cultivars were analyzed to identify candidate genes and develop molecular markers for seed oil content across multiple environments. Transcriptome analysis identified 5458 differentially expressed genes (DEGs) including 2243 positive DEGs and 3215 negative DEGs involved in oil synthesis process. Genome-wide association study identified 48 significant insertion/deletion (InDel) markers associated with seed oil content across five environments. A comparative genomics and transcriptomics analysis detected a total of 147 common gene clusters located in 17 chromosomes. Interestingly, an InDel cluster associated with seed oil content on A03 chromosome was detected in three different environments. Candidate genes identified on A03 form a haplotype, in which variable alleles were found to be different in oil content in an independent population. This locus is important for understanding the genetic control of peanut oil content and may be useful for marker-assisted selection in peanut breeding programs.
Similar content being viewed by others

Introduction
Oil crops are the major source of edible oils which are essential part of human food chain and their health. Peanut, also known as groundnut (Arachis hypogaea L.; AABB, 2n = 4× = 40), is one of the most important oil crops worldwide, with an average oil content of ~ 50% w/w (Barrientos-Priego et al. 2002; Yol et al. 2017). Peanut also has been recognized as a functional food due to a well-balanced fatty acid profile and high levels of specific antioxidants and mono- and polyunsaturated fatty acids (Akhtar et al. 2013). Despite the typically high levels of oil content in currently grown peanut varieties, still there is significant variation among germplasm, i.e., ranging from 31.7 to 57.0% w/w (Yol et al. 2017) which can be used for further enhancing the oil content in newly developed varieties. Because of the above reasons, several breeding programs across the world, including in China, are engaged in developing peanut cultivars with high oil content (Barrientos-Priego et al. 2002; Chen et al. 2016). Despite being such an important trait for several oilseed crops, mere understanding on biological pathway for triacylglycerol (the major form of storage oil in oilseeds) synthesis could be developed (Baud and Lepiniec 2010; Li-Beisson et al. 2013) while the genetic and molecular mechanisms underlying variation in seed oil content of oil crops, including peanut, still remain poorly understood. Improved understanding on genomic control and deployment of genomics tools for several traits successfully accelerated the process and precision in enhancing the genetic gains by developing improved peanut varieties with high trait value (Pandey et al. 2012; Varshney et al. 2013; Pandey et al. 2016; Pandey et al. 2018). Therefore, development and deployment of genomic tools for high oil content will have wider implications in developing improved peanut varieties with enhanced oil content across the globe including China.
Bi-parental populations have been used widely in previous studies to dissect the genetic architecture of complex traits in a number of species followed by discovery of quantitative trait loci (QTLs), candidate genes, and linked markers. Many of these studies have focused on oil content which appears to be a polygenic and quantitative trait due to interactions between multiple genes and the environment. For instance, based on bi-parental linkage mapping, a number of QTL associated with oil content have been identified in all 19 linkage groups of Brassica napus, another globally important oil crop (Delourme et al. 2006; Sun et al. 2012; Zhao et al. 2012; Jiang et al. 2014). Further to this, 11 QTLs associated with oil content in soybean have been detected (Eskandari et al. 2013), and 11 QTLs associated with maize kernel oil concentration were detected across nine chromosomes (Yang et al. 2016). In peanut, 6 and 9 QTLs controlling oil content were respectively identified in two different peanut recombinant inbred line (RIL) populations (Pandey et al. 2014a) and 15 QTLs identified in two or more environments were present across environments related to fatty acid concentrations (Wilson et al. 2017). The speedy progress in sequencing technologies reduced the cost of sequencing as well as sequencing-based genotyping which facilitated feasibility of performing genome-wide association studies (GWAS) in addition to alternative to genetic mapping. In contrast to genetic mapping, the GWAS offers a higher-resolution mapping to detect marker-trait associations followed by candidate gene discovery compared with bi-parental linkage mapping due to higher levels of genetic recombination across the population ancestry (Xu et al. 2016). The approach has previously been used successfully to elucidate the control of several important traits in different crops such as drought resistance in Oryza sativa (Huang et al. 2010; Courtois et al. 2013), flowering time and leaf architecture in Zea mays (Remington et al. 2001; Tian et al. 2011), oil content in Brassica napus (Liu et al. 2016), and root system architecture for efficient absorption of phosphorus in Brassica napus (Wang et al. 2017).
Cultivated peanut is an allopolyploid species with complex genome structure. There are high levels of homology between the peanut A- and B-subgenome, in which both homologous and non-homologous exchanges have been extensively observed (Bertioli et al. 2016). This complexity imposes a huge challenge for discovery of high-quality molecular markers for use in association studies. So far, the application of association genetics in peanut is rare and only one comprehensive GWAS has been reported for several agronomically important traits (Pandey et al. 2014b). However, several studies have been conducted in other allopolyploid species, indicating the potential application of such techniques in peanut. For instance, Harper et al. (2012) identified genetic loci controlling glucosinolate and erucic acid content using association analyses based on transcriptome data in a population of 53 Brassica napus (Brassica AACC; 2n = 38) genotypes. The use of transcriptome data reduced the complexity typically associated with polyploid genomes, as well as allowing the use of both single nucleotide polymorphism (SNP) markers and gene expression markers (GEMs) by measuring transcript abundance. Further studies using the same techniques in larger populations of 84 and 101 genotypes identified genetic loci associated with the control of anion homeostasis and seed glucosinolate concentration (Koprivova et al. 2014; Lu et al. 2014). Another association study deployed Brassica 60K SNP array on a panel of 472 rapeseed genotypes and identified 24,256 polymorphic SNPs which facilitated detection of a region on chromosome A08 significantly associated with seed oil content (Li et al. 2014). The above examples indicate the ability of association genetics to identify loci controlling such complex traits such as oil content in polyploid organisms.
After SNPs, insertions and deletions (InDels), originated from replication slippage, transposable elements, and crossing-over, are the second most abundant structural variations in plant genomes including peanut. These markers have an array of applications including genetic diversity, trait mapping, and molecular breeding. Therefore, in the present study, we developed and deployed a total of 61,942 insertion–deletion (InDel) markers from the transcriptome of peanut pods for conducting GWAS to identify genetic loci associated with oil content. The genomic loci and candidate genes identified in this study have improved understanding on genomic control for high oil content in addition to possibility of developing user-friendly genetic markers for use in molecular breeding for enhancing the level of oil content in improved peanut varieties.
Materials and Methods
Plant Materials
The association panel comprised of 49 peanut accessions which were collected from major breeding centers located in China, i.e., 28 lines from Shandong; 5 from Henan; 4 from Sichuan; 3 from Guangdong; 2 from Hubei;1 each from Hebei, Fujian, and Shanxi; 3 from the USA; and 1 from Mali (Supplementary Table 1).
Field Experiments and Phenotyping
All the 49 genotypes were planted at Laixi, China (120.53° E, 36.86° N), in 2015–2016 and at Qingdao, China (120.41° E, 36.39° N), in 2014–2015 and 2015–2016 and in greenhouse in 2015–2016 at Qingdao. All the field experiments followed a randomized block design with three replicates. Twelve individual plants were planted in a single row for each accession. The trial management followed standard breeding field protocols. At maturity stage, five representative plants from the center of each plot were harvested. The oil content of the desiccated seeds was determined by Soxhlet extraction (ISO 659:2009), and the “oil content” w, expressed as percentage by mass of the product as received, is given by the following:
where
- p :
-
is the percentage, by mass, of total fines;
- I 0 :
-
is the percentage, by mass, of oleaginous impurities;
- I n :
-
is the percentage, by mass, of non-oleaginous impurities;
- w 1 :
-
is the percentage, by mass, of oil in the pure seeds; and
- w 2 :
-
is the percentage, by mass, of oil in the impurities.
Calculation of the Best Linear Unbiased Prediction of Oil Content
As the oil content of the peanut association panel was investigated in multiple environments at multiple time points with multiple replications, the best linear unbiased prediction (BLUP) of oil content for each line across the four environments was calculated using an R script (www.eXtension.org/pages/61006, 20 November 2015, date last accessed) based on a linear model. Means (BLUP) were estimated using the (genotype) term as a fixed factor, retaining [(replicate/row/location/year)] as random factors. Random terms and no defined fixed factor were used to estimate sources of variation. These data were handled separately in an additional GWAS.
Genotyping of the Association Panel and In Silico Mapping of Markers
The 30–40 days immature pods (stages 2–3) from each of the 49 accessions were sampled as explained in Lynch and Wilson (1991). Samples were cleaned and immediately placed in liquid nitrogen before being stored at − 80 °C (Wei et al. 2016). The sequencing libraries of 147 RNA samples were generated using the Illumina RNA Library Prep Kit (NEB #E7760, San Diego, CA, USA) and sequenced on an Illumina Hiseq 2000 platform with 100-bp paired-end reads. Sequencing reads were aligned to the peanut “pseudomolecules” reference genome (AA, Arachis duranensis; BB, Arachis ipaensis, https://www.peanutbase.org/home) with Bowtie2 (Langmead et al. 2012) and then assembled using TopHat 2.0.0 and Cufflinks (Trapnell et al. 2012).
The InDel markers detected by GATK software (https://gatkforums.broadinstitute.org/gatk/) with call frequencies missing < 20% and minor allele frequencies (MAF) > 0.05 were selected for association mapping analysis. The physical position of the markers was identified by aligning the sequence of a 100-bp paired-end reads attached to each marker with the “pseudomolecules” genome sequences of peanut (Arachis duranensis—AA and Arachis ipaensis—BB) using local BLASTn (BLAST, Basic Local Alignment Search Tool; http://blast.ncbi.nlm.nih.gov/Blast.cgi). If the reads matched two or more locations in the reference genome of peanut, the markers were regarded as non-specific markers and discarded.
Population Structure and Relative Kinship Analysis of the Association Panel
Genotype–phenotype covariance can lead to spurious associations in GWAS due to unknown ancestry (Lander and Schork 1994). Population structure and kinship can be estimated and used in linear models to correct for such false associations. Using the Bayesian Markov chain Monte Carlo (MCMC) model in STRUCTURE V. 2.3.4 (Xu et al. 2016; Wang et al. 2017), the population structure of the peanut association panel was estimated. Based on a model for admixture and correlated allele frequencies in STRUCTURE V.2.3.4, a population parameter (K) value was obtained across five iterative runs with a putative number of populations set from 1 to 10. The length of the burn-in period and number of MCMC replications after burn-in were each set to 100,000. After calculating the rate of change in log probability of the data (LnP(D)) and an ad hoc statistic ΔK between successive K values, the true K value was determined (Evanno et al. 2005). A relative kinship matrix was calculated by SPAGeDi (http://ebe.ulb.ac.be/ebe/SPAGeDi.html) with all negative kinship values between two individuals set to 0 (Wang et al. 2017).
Genome-Wide Association Analysis of Oil Content Traits
Population structure and kinship could be used for correcting the spurious association created by genotype–phenotype covariance. Generalized linear model (GLM) and mixed linear model (MLM) calculations were performed in TASSEL version 5.0 to determine the best model for association analyses. The GLM included a naïve model without controlling for population structure and a Q model which controlled for population structure using a Q matrix to identify subpopulation fit as covariate in the general linear model. The MLM included a K model which assesses pair-wise relationship between the individuals using the kinship matrix obtained in SPAGeDi software and a Q+K model which controlled for population structure using subpopulation and kinship between the genotypes. Quantile–quantile plots were created with a negative log10(P) value of the expected P value from the genotype–phenotype association and the expected P value under the assumption that no association exists between genotype and phenotype. The threshold of significance was set to P < 1.61 × 10−5 (1/total markers used) (Wei et al. 2016). The phenotype or trait was determined by GWAS, and the trait y is given by the following (Yu et al. 2006):
where
- y :
-
is the trait values,
- α :
-
is the environments,
- σ :
-
is the candidate markers effects,
- γ :
-
is the subpopulation effects, and
- δ :
-
is the background genetic effects.
Differentially Expressed Genes Underlying Significant SNP Loci and Favorable Alleles for Seed Oil Content
Three high oil content cultivars and three low oil content cultivars selected from the panel of 49 cultivars for differentially expressed gene (DEG) analysis. Differentially expressed genes between high and low were identified based on criteria FDR < 0.01 and |log2 (FPKMhigh)/FPKMlow| > 2. Based on physical map and gene annotation, the genes located in the LD region of the significant InDel markers “InDel6418” were identified at three different environments (Oilcontent2016-QD2, Oilcontent2016-QD1, and OilContent-BLUP). Due to an inconsistent number of SNPs for each InDel marker, SNP markers in a locus associated with peanut oil content on chromosome were used to analyze the relationship between markers haplotypes and phenotypes in the association panel. More than three cultivars with the same SNP defined a haplotype. The cultivar phenotypes were figured by Origin 8 software.
Results
Phenotypic Variation of Peanut Seed Oil Content
Extensive phenotypic variation was observed in oil content within and across different environments. Across the five environments, the seed oil content of the peanut association panel showed continuous variation and approximated a normal distribution (Supplementary Fig. 1). Peanut seed oil content ranged from 40.01 to 58.11%, with an average of 48.10% in 2016 at Qingdao2 environment; from 44.35 to 59.34%, with an average of 52.74% in 2016 at Qingdao1 environment; from 43.66 to 57.86%, with an average of 50.41% in 2016 at Laixi environment; and from 46.40 to 58.00%, with an average of 50.60% in 2015 at Qingdao environment (Table 1). The oil content coefficient of variation ranged from 5.37 to 9.47%, with an average of 6.93%, suggesting that the oil content of most genotypes is stable in different environments and relatively insensitive to environmental factors.
Population Structure and Relative Kinship
A total of 61,942 InDels with MAF > 0.05 and call frequencies > 0.8 were matched to a unique position in the “pseudomolecules” of diploid genome sequences of peanut progenitors (Arachis duranensis—AA and Arachis ipaensis—BB) (Supplementary Table 2). These InDels were used to assess the population structure and relative kinship of the association panel, in addition to association analyses. A total of 61,942 InDel markers were identified across all chromosomes. InDel number on each chromosome ranged from 2069 on A07 to 4724 on B03. The density of InDel markers on each chromosome ranged from 46.74 kb/InDel on B08 to 17.96 kb/InDel on A08 (Table 2).
The K values obtained through population structure in the peanut association panel increased continuously with no obvious inflection points of LnP(K) values (Fig. 1a). The ∆K values indicated that the rate of change ad hoc statistic was equal to 2; therefore, the population could be divided into two subgroups: subgroup 1 and subgroup 2 (Supplementary Table 1). Of the four lines grouped together in subgroup 1, three originated from Shandong while single lines were from Guangzhou. Subgroup 2 was big with 45 lines originated from Shandong, Fujian, Guangdong, Hebei, Henan, Hubei, Shanxi, Sichuan, the USA, and Mali (Supplementary Table 1). The relative kinship between genotypes in the association panel analysis showed that the genotypes with kinship coefficients less than 0.1 accounted for 79.77% of the association panel (Fig. 1b), indicating that the majority of the genotypes have a weak relationship with one another.

The population structure and relative kinship of the peanut panel. a The rate of change in log probability of the data (LnP(D)) and ad hoc statistic ΔK (delta K) of population structure in the 49 peanut population association panel. b The relative kinship of the peanut panel
Genome-Wide Association Analysis and Gene Expression Profile of Candidate Genes
The Naïve model, Q model, K model, and Q+K model were used for association mapping. The deviations of observed values from the expected values are shown in QQ plots (Supplementary Fig. 2) and were applied to select the most suitable model for each trait in the different experiments (Fig. 2a–e). Based on the results of QQ plot, Q+K, K, K, GLM, and K models are fitful for 2015Qingdao, 2016Laixi, 2016Qingdao1, 2016Qingdao2, and mean (BLUP) environments, respectively. A total number of 19, 5, 10, 8, and 6 significant InDel markers for oil content were detected in 2015Qingdao, 2016Laixi, 2016Qingdao1, 2016Qingdao2 and mean (BLUP) environments, respectively (Fig. 2; Supplementary Table 3). The GWAS analysis identified a total of 48 significant loci for oil content in five different environments. The significant loci were located across 18 chromosomes (Fig. 3; Supplementary Table 3). One of the significant loci (InDel6418) located on A03 chromosome was detected across three different environments (Fig. 4). This locus had shown high phenotypic variation explained (PVE, nearly 30%) for oil content indicating its importance for further investigation on its role in oil biosynthesis mechanism and use in molecular breeding.

Genome-wide association scanned for oil content of peanut. a Manhattan and optimal quantile–quantile plot for oil content detected in 2016QD2 environment. b Manhattan and optimal quantile–quantile plot for oil content detected in 2016QD1 environment. c Manhattan and optimal quantile–quantile plot for oil content using the BLUP value. d Manhattan and optimal quantile–quantile plot for oil content detected in 2015QD environment. e Manhattan and optimal quantile–quantile plot for oil content detected in 2016LX environment

The distribution pattern of 147 candidate differential expression genes (DEGs) and their corresponding InDels associated with oil content. InDels and candidate genes are marked in black and red, respectively. Positive and negative DEGs are marked in red blocks and blue blocks

Co-localized loci on chromosome A03 for oil content of peanut. a Significant InDel associated with oil content in 2016QD1 environment. b Significant InDel associated with oil content in 2016QD2 environment. c Significant InDel associated with oil content using the BLUP value. d Expression profile detected by transcriptome sequencing of candidate genes located in the LD intervals
After ranking the genotypes in the association panel by oil content (Supplementary Fig. 3), three high (31, 44, 46) and three low (7, 8, 14) oil content genotypes were selected for expression profiling of candidate genes related to oil content (Supplementary Table 4). A total of 5458 differently expressed genes were identified between high and low oil content genotypes (Supplementary Table 5). A comparison of our GWAS and transcriptome sequencing results revealed 147 common gene clusters located in 17 chromosomes in peanut (Supplementary Fig. 3). Genes at these loci may be involved in the control of peanut oil content and are worthy for further investigation.
Haplotypes Associated with Peanut Oil Content Traits
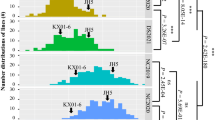
On chromosome A03, 34 genes were housed in the candidate genomic interval which is home for the significant InDel6418 (CTTTTTT/-) locus (Fig. 4d) detected at three different environments (Oilcontent2016-QD2, Oilcontent2016-QD1, and OilContent-BLUP). Two genes (LOC107478374 and LC10747828) were located in the interval region of the peak marker InDel6418 at 24.15–26.15 Mb. There were ten SNPs located in this interval (Fig. 5f). Five of ten SNPs formed a region with five different haplotypes on A03 chromosome. The cultivars or inbred lines with haplotype 2 (CTTGA, n = 13) had higher average peanut oil content (51.26 ± 4.44%) than those with other haplotypes in five environments (Fig. 5a–e), whereas the varieties with haplotype 3 (YTTGA, n = 5) had lower average peanut oil content (49.64 ± 3.20%) than those with the other haplotypes (Fig. 5a–e).

The haplotypes associated with oil content traits in the five different environments. a The haplotypes associated with oil content in 2015QD environment. b Oil content in 2016QD1 environment. c Oil content in 2016QD2 environment. d Oil content in 2016LX environment. e Oil content using the BLUP value. f The haplotypes on A03 (red color trilateral)
Discussion
Development of peanut varieties with high oil content is the most important breeding goals after pod yield for peanut improvement programs globally. Detection of genetic loci related to oil content could guide such breeding efforts in more fast and efficient manner. Previous studies based on bi-parental linkage mapping for seed oil content have identified ten main-effect QTLs for oil content and oil quality traits of peanut (Pandey et al. 2014b). Fifteen QTLs identified in two or more environments that were present across environments related to fatty acid concentrations were detected in peanut BC3F6 population (Wilson et al. 2017). GWAS performed with a large number of SNPs and InDels has been reported in many plants and crops, such as Arabidopsis (Atwell et al. 2010), rice (Huang et al. 2010), and maize (Kump et al. 2011; Li et al. 2013). There were many oil content studies reported in recent years where single locus (BnaA.FAE) to 50 loci significantly associated with seed oil content were identified in B. napus (Li et al. 2014; Liu et al. 2016). In this study, 48 loci significantly associated with peanut oil content were detected across multiple environments. A total of 6 of 48 significant loci were located in more than two different environments (Supplementary Table 3).
There are many elements that affect the power of GWAS such as marker density, population size, and statistical methods. Markers can be obtained by the advances in genome sequencing such as reference genome sequences of diploid progenitors of cultivated tetraploid (Bertioli et al. 2016) and 58K SNP arrays (Pandey et al. 2017) in peanut. In the present study, we have successfully identified > 60K InDels and 20K SNP markers through mRNA-seq (Supplementary Table 2; Supplementary Table 4). Population structure and relative kinship were used for controlling false-positive results in GWAS. The association panel was classified into two subpopulations (Fig. 1a, c) in our studies, co-incidentally similar to studies conducted with elite varieties in rice (Huang et al. 2010) and rapeseed (Xu et al. 2016; Liu et al. 2016; Wang et al. 2017). The K matrix (unequal relatedness among individuals) was the main elements of MLM statistical models. In the present study, genotypes with kinship coefficients less than 0.1 accounted for 79.77% of the association panel (Fig. 1b), indicating that the genotypes in the association panel were only weakly related. Four different models were constructed for controlling spurious associations, such as the naïve model, Q model, K model, and Q+K model (Supplementary Fig. 2). When kinship is ignored in our association study, the P values were found to be significantly inflated, indicating that relative kinship within the population is likely to be one of the major factors resulting in false positives (Supplementary Fig. 2). With the addition of kinship in the models, such false positives are controlled in the results obtained in the present investigation.
Oil content is one of most important agricultural traits in peanut. Previous studies on the high oil content of peanut were mainly conducted using QTL mapping (Pandey et al. 2014b). However, GWAS were implemented in a collection of peanut germplasms in our studies (Supplementary Table 1). Oil content is a complex trait and our investigation identified 5458 DEGs including 2243 positive DEGs and 3215 negative DEGs involved in the oil synthesis process (Fig. 3; Supplementary Table 5). Breeders have matched functionally different oil-related alleles to specific environments during the peanut breeding process (Liu et al. 2016). In this study, the association panel of 49 genotypes of peanut was planted in five different environments and the seed oil content quantified (Supplementary Fig. 3). Five SNPs near a significant InDel marker on chromosome A03 formed a haplotype which was shown to contribute a major percentage of peanut oil content (Fig. 5). Favorable alleles at this locus may be an effective way to increase the oil content during the selection of high oil lines using molecular breeding. The discovery of an optimal haplotype on A03 provides a useful resource which may enable accurate selection of peanut with higher seed oil content and improve our understanding of the molecular mechanisms related to oil accumulation in plants.
References
Akhtar S, Khalid N, Ahmed I, Shahzad A, Suleria HAR (2013) Physicochemical characteristics, functional properties, and nutritional benefits of peanut oil: a review. Crit Rev Food Sci Nutr 54:1562–1575
Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JDG, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631
Barrientos-Priego L, Isleib TG, Pattee HE (2002) Variation in oil content among Mexican and Peruvian hirsuta peanut landraces and Virginia-type hypogaea lines. Peanut Sci 29:72–77
Baud S, Lepiniec L (2010) Physiological and developmental regulation of seed oil production. Prog Lipid Res 49:235–249
Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EK et al (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 48:438–446
Chen X, Li H, Pandey MK, Yang Q, Wang X, Garg V, Li H, Chi X, Doddamani D, Hong Y, Upadhyaya H, Guo H, Khan AW, Zhu F, Zhang X, Pan L, Pierce GJ, Zhou G, Krishnamohan KAVS, Chen M, Zhong N, Agarwal G, Li S, Chitikineni A, Zhang GQ, Sharma S, Chen N, Liu H, Janila P, Li S, Wang M, Wang T, Sun J, Li X, Li C, Wang M, Yu L, Wen S, Singh S, Yang Z, Zhao J, Zhang C, Yu Y, Bi J, Zhang X, Liu ZJ, Paterson AH, Wang S, Liang X, Varshney RK, Yu S (2016) Draft genome of the peanut A-genome progenitor (Arachisduranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc Natl Acad Sci U S A 113:6785–6790
Courtois B, Audebert A, Dardou A, Roques S, Ghneim-Herrera T, Droc G et al (2013) Genome-wide association mapping of root traits in a japonica rice panel. PLoS One 8:e78037
Delourme R, Falentin C, Huteau V, Clouet V, Horvais R, Gandon B, Specel S, Hanneton L, Dheu JE, Deschamps M, Margale E, Vincourt P, Renard M (2006) Genetic control of oil content in oilseed rape (Brassica napus L). Theor Appl Genet 113:1331–1345
Eskandari M, Cober ER, Rajcan I (2013) Genetic control of soybean seed oil: II QTL and genes that increase oil concentration without decreasing protein or with increased seed yield. Theor Appl Genet 126:1677–1687
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Harper AL, Trick M, Higgins J, Fraser F, Clissold L, Wells R, Hattori C, Werner P, Bancroft I (2012) Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat Biotechnol 30:798–802
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Li W, Lin Z, Buckler ES, Qian Q, Zhang QF, Li J, Han B (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967
ISO (2009) ISO “659: 2009, oilseeds—determination of oil content”. International Organization for Standardization, Geneva
Jiang C, Shi J, Li R, Long Y, Wang H, Li D, Zhao J, Meng J (2014) Quantitative trait loci that control the oil content variation of rapeseed (Brassica napus L.). Theor Appl Genet 127:957–968
Koprivova A, Harper AL, Trick M, Bancroft I, Kopriva S (2014) Dissection of the control of anion homeostasis by associative transcriptomics in Brassica napus. Plant Physiol 166:442–627
Kump KL, Bradbury PJ, Wisser RJ, Buckler ES, Belcher AR, Oropeza-Rosas MA, Zwonitzer JC, Kresovich S, McMullen MD, Ware D, Balint-Kurti PJ, Holland JB (2011) Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet 43:163–168
Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li H, Peng Z, Yang X, Wang W, Fu J, Wang J, Han Y, Chai Y, Guo T, Yang N, Liu J, Warburton ML, Cheng Y, Hao X, Zhang P, Zhao J, Liu Y, Wang G, Li J, Yan J (2013) Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet 45:43–50
Li F, Chen B, Xu K, Wu J, Song W, Bancroft I, Harper AL, Trick M, Liu S, Gao G, Wang N, Yan G, Qiao J, Li J, Li H, Xiao X, Zhang T, Wu X (2014) Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L). DNA Res 21:355–367
Li-Beisson Y, Shorrosh B, Beisson F, Andersson MX, Arondel V, Bates PD, Baud S, Bird D, DeBono A, Durrett TP, Franke RB, Graham IA, Katayama K, Kelly AA, Larson T, Markham JE, Miquel M, Molina I, Nishida I, Rowland O, Samuels L, Schmid KM, Wada H, Welti R, Xu C, Zallot R, Ohlrogge J (2013) Acyl-lipid metabolism. Arabidopsis Book 11:e0161
Liu S, Fan C, Li J, Cai G, Yang Q, Wu J, Yi X, Zhang C, Zhou Y (2016) A genome wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor Appl Genet 129:1203–1215
Lu GY, Harper AL, Trick M, Morgan C, Fraser F, O’Neill C, Bancroft I (2014) Associative transcriptomics study dissects the genetic architecture of seed glucosinolate content in Brassica napus. DNA Res 21:613–625
Lynch RE, Wilson DM (1991) Enhanced infection of peanut, Arachis hypogaea L., seeds with Aspergillus flavus group fungi due to external scarification of peanut pods by the lesser cornstalk borer, Elasmopalpus lignosellus (Zeller). Peanut Science 18:110–116
Pandey MK, Monyo E, Ozias-Akins P, Liang X, Guimarães P, Nigam SN, Upadhyaya HD, Janila P, Zhang X, Guo B, Cook DR, Bertioli DJ, Michelmore R, Varshney RK (2012) Advances in Arachis genomics for peanut improvement. Biotechnol Adv 30(3):639–651
Pandey MK, Wang ML, Qiao L, Feng S, Khera P, Wang H, Tonnis B, Barkley NA, Wang J, Holbrook CC, Culbreath AK, Varshney RK, Guo B (2014a) Identification of QTLs associated with oil content and mapping FAD2 genes and their relative contribution to oil quality in peanut (Arachis hypogaea L). BMC Genet 15:133
Pandey MK, Upadhyaya HD, Rathore A, Vadez V, Sheshshayee MS, Sriswathi M et al (2014b) Genome-wide association studies for 50 agronomic traits in peanut using the ‘reference set’ comprising 300 genotypes from 48 countries of semi-arid tropics of the world. PLoS One 9(11):e113326
Pandey MK, Roorkiwal M, Singh V, Lingam A, Kudapa H, Thudi M, Chitikineni A, Rathore A, Varshney RK (2016) Emerging genomic tools for legume breeding: current status and future perspectives. Front Plant Sci 7:455
Pandey MK, Agarwal G, Kale SM, Clevenger J, Nayak SN, Sriswathi M, Chitikineni A, Chavarro C, Chen X, Upadhyaya HD, Vishwakarma MK, Leal-Bertioli S, Liang X, Bertioli DJ, Guo B, Jackson SA, Ozias-Akins P, Varshney RK (2017) Development and evaluation of a high density genotyping ‘Axiom_Arachis’ array with 58K SNPs for accelerating genetics and breeding in groundnut. Sci Rep 7:40577
Pandey MK, Singh VK, Roorkiwal M, Thudi M, Saxena RK, Varshney RK (2018) Sequencing-based next generation breeding for legume improvement. In: Rao PV, Muralidharan K, Siddiq EA (eds) Modern breeding strategies for crop improvement. Professor Jayashankar Telangana State Agricultural University (PJTSAU), Rajendranagar, ISBN: ISBN 978-81-936934-0-7, pp 100–119
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci U S A 98:11479–11484
Sun M, Hua W, Liu J, Huang S, Wang X, Liu G, Wang H (2012) Design of new genome- and gene-sourced primers and identification of QTL for seed oil content in a specially high-oil Brassica napus cultivar. PLoS One 7:e47037
Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43:159–162
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578
Varshney RK, Mohan SM, Gaur PM, Gangarao NVPR, Pandey MK, Bohra A, Sawargaonkar SL, Chitikineni A, Kimurto PK, Janila P, Saxena KB, Fikre A, Sharma M, Rathore A, Pratap A, Tripathi S, Datta S, Chaturvedi SK, Mallikarjuna N, Anuradha G, Babbar A, Choudhary AK, Mhase MB, Bharadwaj C, Mannur DM, Harer PN, Guo B, Liang X, Nadarajan N, Gowda CLL (2013) Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol Adv 31:1120–1134
Wang X, Chen Y, Thomas CL, Ding G, Xu P, Shi D, Grandke F, Jin K, Cai H, Xu F, Yi B, Broadley MR, Shi L (2017) Genetic variants associated with the root system architecture of oilseed rape (Brassica napus L) under contrasting phosphate supply. DNA Res 24(4):407–417. https://doi.org/10.1093/dnares/dsx013
Wei L, Jian H, Lu K, Filardo F, Yin N, Liu L, Qu C, Li W, Du H, Li J (2016) Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol J 14:1368–1380
Wilson JN, Chopra R, Baring MR, Selvaraj MG, Simpson CE, Chagoya J, Burow MD (2017) Advanced backcross quantitative trait loci (QTL) analysis of oil concentration and oil quality traits in peanut (Arachis hypogaea L). Trop Plant Biol 10:1–17
Xu L, Hu K, Zhang Z, Guan C, Chen S, Hua W, Li J, Wen J, Yi B, Shen J, Ma C, Tu J, Fu T (2016) Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L). DNA Res 23:43–52
Yang Z, Li X, Zhang N, Zhang YN, Jiang H W, Gao J, Kuai BK, Ding YL, Huang XQ (2016) Detection of quantitative trait loci for kernel oil and protein concentration in a B73 and Zheng58 maize cross. Genet Mol Res 15(3). https://doi.org/10.4238/gmr.15038951
Yol E, Ustun R, Golukcu M, Uzun B (2017) Oil content, oil yield and fatty acid profile of groundnut germplasm in mediterranean climates. J Am Oil Chem Soc 94:787–804
Yu J, Pressoir G, Briggs WH, Vroh BI, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38(2):203–208
Zhao J, Huang J, Chen F, Xu F, Ni X, Xu H, Wang Y, Jiang C, Wang H, Xu A, Huang R, Li D, Meng J (2012) Molecular mapping of Arabidopsis thaliana lipid-related orthologous genes in Brassica napus. Theor Appl Genet 124:407–421
Funding
This work was supported by the grants from the National High Technology Research and Development Program of China (No.2013AA102602); the Agricultural Scientific And Technological Innovation Project of Shandong Academy of Agricultural Sciences (No.CXGC2016B02); the National Natural Science Foundation of China (No.31471533); the National Ten Thousand Youth Talents Plan of 2014 (No. W02070268); and the China Agriculture Research System (No.CARS-13). The work reported in this article was undertaken as a part of the CGIAR Research Program on Grain Legumes and Dryland Cereals (GLDC). ICRISAT is a member of the CGIAR.
Author information
Authors and Affiliations
Contributions
MY conceived the study. XW, PX, and MY designed the experiments. YL measured oil contents of accessions panel. LY, YR, YS, and LL organized the implementation of field trials. QX, PX, and XW analyzed the data. XW, PX, and MY wrote the paper. LY, TDA, WQ, XC, QX, MKP, RKV, and MY participated in the interpretation of results and modification of the manuscript. All the authors have read and approved the publication of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, X., Xu, P., Yin, L. et al. Genomic and Transcriptomic Analysis Identified Gene Clusters and Candidate Genes for Oil Content in Peanut (Arachis hypogaea L.). Plant Mol Biol Rep 36, 518–529 (2018). https://doi.org/10.1007/s11105-018-1088-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-018-1088-9