Abstract
Systematic evolution of ligands by exponential enrichment (SELEX) offers a powerful method to isolate affinity oligonucleotides known as aptamers, which can then be used in a wide range of applications from drug delivery to biosensing. However, conventional SELEX methods rely on labor intensive and time consuming benchtop operations. A simplified microfluidic approach is presented which allows integration of the affinity selection and amplification stages of SELEX for the isolation of target-binding oligonucleotides by combining bead-based biochemical reactions with free solution electrokinetic oligonucleotide transfer. Free solution electrokinetics allows coupling of affinity selection and amplification for closed loop oligonucleotide enrichment without the need for offline processes, flow handling components or gel components, while bead based selection and amplification allow efficient manipulation of reagents and reaction products thereby realizing on-chip loop closure and integration of the entire SELEX process. Thus the approach is capable of multi-round enrichment of oligonucleotides using simple transfer processes while maintaining a high level of device integration, as demonstrated by the isolation of an aptamer pool against a protein target (IgA) with significantly higher binding affinity than the starting library in approximately 4 hours of processing time.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Aptamers are single-stranded oligonucleotides, including single-stranded deoxyribonucleic acid (DNA), ribonucleic acid (RNA) and peptide molecules, which can be used for specific molecular recognition toward a broad spectrum of targets including small molecules,1 peptides,2 amino acids,3,4 proteins,5,6 cells,7,8 viruses,9,10 and bacteria.11 In many applications aptamers can be used as substitutes to antibodies but with the additional unique advantages of reversible binding, long shelf life, low batch-to-batch variability,12 low immunogenicity,13 long-term stability, ease of coupling to other functional molecular groups,14,15 and ability to be discovered through an in vitro process.16,17 Thus, aptamers are being employed as biosensors, drug delivery devices, and bio-imaging probes in many applications within basic biological sciences,18 drug discovery,19 and clinical diagnostics and therapeutics.16,20
The in vitro process used to discover aptamers with high affinity for a particular target is known as systematic evolution of ligands by exponential enrichment (SELEX). During the SELEX process an aptamer is found from an initial library of randomized combinatorial oligonucleotides with approximately 1012–1015 unique members through iterative rounds of affinity selection and amplification.17,21,22 Affinity selection involves incubating the library with a target of interest and then separating library members that bind to the target from those that do not bind to the target. The target-binding oligonucleotides are then amplified through the polymerase chain reaction (PCR) during the amplification stage and converted back to single-stranded oligonucleotides. These amplified single-stranded oligonucleotides are then incubated with the target as the affinity selection of a new round SELEX begins. Therefore, in every subsequent round of selection the oligonucleotides must compete for binding from the oligonucleotides that bound in the previous round. These rounds of affinity selection and amplification are repeated until a pool of oligonucleotides is isolated with high affinity and specificity toward the target. While effective, this process conventionally involves extensive manual handling of reagents, and is time consuming, typically requiring a month or more to complete a single SELEX run.23
Microfluidic technology has been recently applied to the SELEX field to rapidly isolate aptamers at low cost in a highly integrated and automated fashion, and additionally with reduced reagent consumption and in fewer rounds of iteration. Microfluidic affinity selection has been performed, in conjunction with off-chip amplification, against targets retained using silica capillary walls,24 microbeads25–32 or sol-gels,33–35 or against cells in solution,36,37 to increase selection stringency,25–29,36,37 create more favorable biomolecular environments,33–35 or allow simultaneous positive and negative selections.38 Moreover, microchips have been developed in an attempt to integrate the overall SELEX process using pneumatically based flow control for isolation of aptamers against viruses,39 cells28,40,41 and proteins.30–32,42,43 While capable of isolating aptamers these microfluidic devices have relied on highly complex fabrication techniques not available to most researchers, extensive flow control components (integrated pumps, valves and mixers which all require additional off-chip equipment), sophisticated operating equipment, complicated designs, and integration of unstable gels.
This paper presents a simplified microfluidic approach using free solution electrokinetic flow control to fully integrate the SELEX process on a single microchip. In this approach, electrophoretic transport of oligonucleotides occurs in free solution between selection and amplification chambers through a microchannel that is highly resistant to bulk flow and diffusion due to a relatively large length and small cross-sectional area. This high resistance prevents cross contamination between the chambers without significantly impeding electrophoretic oligonucleotide transport. The design hence obviates the use of complex flow handling or gel preparation, thereby allowing simple and efficient transfer of target-binding oligonucleotides as well as a high level of device integration. Also, the device is fabricated without the need for the multi-layer soft lithography techniques previously reported devices have required. These advantages are combined with a bead-based protocol to achieve on-chip coupling of affinity selection and amplification of target-binding oligonucleotides without requiring offline processes. The device is further simplified, while the reliability is improved, through optimized design of the microfluidic geometry and use of a magnetic microbead retention scheme that make the device considerably less prone to potentially catastrophic trapped bubbles.44 The practical utility of the device is demonstrated with experimental results on isolation of DNA aptamers against human immunoglobulin A (IgA) as a representative target.
Experimental
Aptamer selection principle
The approach involves iterative rounds of microbead-based affinity selection and PCR amplification coupled by electrokinetic fluidic handling (Figure 1) carried out on a microchip. Aptamers are enriched from an initial single-stranded DNA (ssDNA) library comprised of oligonucleotides with a randomized region flanked by a forward primer region on the 5' end, and a region defined to be complementary to the reverse primer on the 3' end. This initial library is incubated with microbeads functionalized with biomolecular target (selection beads) during affinity selection. Through multiple buffer washes non-binding and weakly binding oligonucleotides are removed and discarded while strongly bound oligonucleotides remain attached to the target-functionalized microbeads. These strong binding oligonucleotides are thermally eluted from the target-functionalized microbeads and electrokinetically transferred to a second set of microbeads (amplification beads) which have been modified with reverse primers thus causing the library to hybridize to the reverse primers tethered to the amplification bead surfaces. PCR reagents are then introduced and PCR thermal cycling commences resulting in amplified copies of the target-binding oligonucleotides hybridized to the bead surfaces. The amplified product is thermally denatured from the bead bound complement and transferred back to replenished selection beads for further rounds of SELEX.

Figure 1. Principle of microfluidic aptamer development: (a) ssDNA with random sequence is introduced to the target. (b) ssDNA is allowed to bind to the target. (c) Weak binding and non-binding oligonucleotides are removed by multiple buffer washes. (d) Strong binders are thermally eluted and (e) transferred to the amplification chamber with electrokinetics. (f) Transferred oligonucleotides hybridize to surface-immobilized reverse primers and (g) amplified through PCR. (h) The amplified single strands are thermally released and (i) electrokinetically transported back to the selection chamber for further affinity selection.
Microfluidic device design and operation
The electrokinetic microfluidic approach for aptamer enrichment is realized in an integrated microchip (Figure 2). The device consists of two (selection and amplification) microchambers of identical geometry (∼2.8-μL) connected by a microchannel. The microchambers are hexagonal in shape with dimensions of 2 mm wide, 7 mm long and 250 um high. The hexagonal chamber shape has been demonstrated to be considerably less prone to trapping bubbles which can cause sample purging at increased temperatures, act as thermal insulators, create uneven temperature distributions, and reduce the effective volume of the chamber.44 Each microchamber features a resistive heater and temperature sensor located beneath the chamber, an inlet, an outlet, and an electrode port. Connecting the two chambers is a single serpentine shaped microchannel (14.75 mm long, 50 μm wide, 20 μm tall) which prevents diffusion between the microchambers but allows oligonucleotide migration under an electric field. Similarly the electrode port (2 mm diameter) is connected to its microchamber through a long, narrow microchannel (4.5 mm long, 50 μm wide, 20 μm tall). The electrode ports allow the insertion of platinum wire electrodes.

Figure 2. (a) Three-dimensional rendering of the microfluidic device. (b) Top view schematic of the microfluidic device. (c) Cross-sectional view of microfluidic device design along line A-A.
Repetitive, closed-loop cycles of affinity selection and PCR amplification can be achieved in the microdevice without the need for offline instruments. First target-functionalized beads are introduced into the selection chamber and retained via an external magnet. Affinity selection then occurs by flowing the initial randomized library across the beads from the inlet to the outlet, allowing oligonucleotides with affinity toward the target to bind to the target-functionalized beads. The target-functionalized beads are washed with flowing buffer to remove weakly binding oligonucleotides. The remaining strongly bound oligonucleotides are then eluted by increasing the chamber temperature with the integrated heater located beneath the chamber. Meanwhile, an electric field is applied from the selection chamber electrode port to the amplification chamber port, inducing migration of the eluted oligonucleotides to the amplification chamber wherein amplification beads are located to capture and immobilize the oligonucleotides in the amplification chamber. PCR reagents are then introduced to the amplification chamber using the amplification chamber inlet and subsequently PCR thermal cycling commences using the integrated heater and temperature sensor located beneath the amplification chamber. The amplified product is thermally released using the integrated heater, an electric field of reversed polarity is applied, and the oligonucleotides are electrokinetically transferred back to the selection chamber where they interact with replenished target-functionalized beads as a new round of affinity selection begins. Note, the resulting amplified product of the PCR is double stranded oligonucleotides consisting of the amplified enriched library and the amplified complement of the enriched library. Partitioning of the desired amplified enriched strands from their complementary strands is achieved directly with the amplification beads. The complementary strand is an extended reverse primer which is coupled to the beads through temperature invariant streptavidin-biotin chemistry while the desired strand is attached to the beads through complementary base pairing to the complementary strand. Thus the desired strand can be released from the bead surfaces (while its complement remains attached to the beads), and subsequently transferred to the selection chamber, with elevated temperatures (∼95°C).
Materials
Human Imunnoglobulin A (IgA1) was purchased from Cell Sciences (Newburyport, MA), and Pierce NHS-activated microbeads were purchased from Thermo Fisher Scientific (Waltham, MA). Ethanolamine, MgCl2, tris, boric acid, and molecular biology grade water were purchased from Sigma-Aldrich (St. Louis, MO). Deoxyribonucleotide triphosphates (dNTPs) and GoTaq Flexi DNA polymerase were obtained from Promega Corp. (Madison, WI). Randomized oligonucleotide library (5' – GAA CTT CGC ATC TCA CGG TGG – 36N – GGA TCC GAT TCC ATC CTG CTC CTC– 3') and primers (Forward Primer: 5' – FAM – GAA CTT CGC ATC TCA CGG TGG -3', and Reverse Primer: 5' – dual biotin – GAG GAG CAG GAT GGA ATC GG – 3') were synthesized and purified by Integrated DNA Technologies (Coralville, IA). Dulbecco's phosphate-buffered saline (D-PBS), and streptavidin coupled magnetic beads (Dynabeads M-270 Streptavidin) were purchased from Invitrogen (Carlsbad, CA). SU-8 (2000, 2025, and 2075 series) negative photoresist was purchased from Microchem Corp. (Westborough, MA). Polydimethylsiloxane (PDMS) was obtained from Robert McKeown Company (Somerville, NJ) and silicon wafers were purchased from Silicon Quest International, Inc. (San Jose, CA).
Magnetic microbeads (Pirece NHS-activated beads) functionalized with a protein were used for selection and magnetic microbeads (Dynabeads M-270 steptavidin) functionalized with reverse primers were used for amplification. The beads for selection were functionalized with a protein (IgA1) through covalent coupling between the NHS groups of the microbeads and the amine groups of the protein. Prior to an experiment, the protein was incubated with IgA1 under gentle rotation for 1 hour at room temperature, and washed with D-PBS buffer. Unreacted NHS groups were passivated by incubating the functionalized beads in ethanolamine. The functionalized beads were then suspended in D-PBS buffer. Similarly, streptavidin magnetic beads were functionalized by incubation with dual biotin conjugated reverse primers through covalent coupling for 30 minutes, washed with D-PBS buffer and suspended in D-PBS buffer.
Fabrication
The microfluidic device was realized in polydimethylsiloxane using soft-lithography techniques (Figure 3). First, a layer of SU-8 was spun, and developed on a silicon wafer to define the narrow channels for oligonucleotide transfer. Then a second, thicker layer of SU-8 was spun, and developed to create the selection and amplification chambers to complete the fabrication of the flow-layer. On a separate glass-slide substrate chrome (10 nm) and gold (100 nm) were thermally evaporated and then patterned and etched to create a glass substrate bearing gold resistive heaters and temperature sensors. A thin layer of PDMS was spun on this glass slide and cured to passivate and allow reuse of the heater and temperature sensors. PDMS was poured on the wafer containing the flow layer features, degassed in a vacuum chamber, and cured at 72°C for 30 minutes. The hardened PDMS was peeled off, cut, and had inlet and outlet holes punched with an autopsy punch. This flow layer was exposed to oxygen plasma along with the glass slide bearing the heater and temperature sensor and the two pieces were bonded and baked at 80°C overnight.
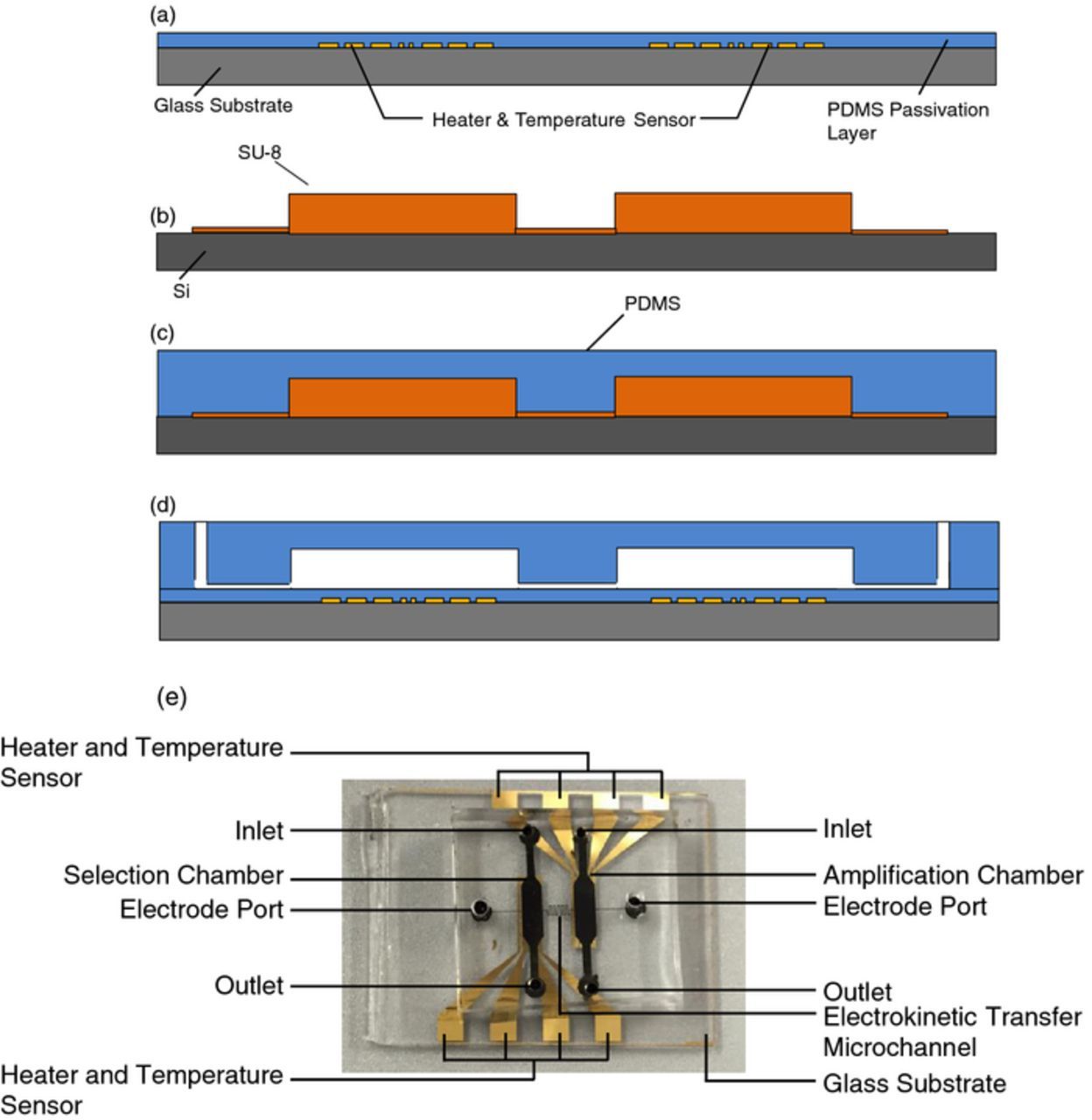
Figure 3. (a) Deposition, patterning and passivation of gold temperature sensors and heaters. (b) Fabrication of the SU-8 mold defining microfluidic chambers and channels. (c) Casting of the PDMS sheet of fluidic chambers and channels. (d) Punching of inlets and outlets and bonding of the PDMS sheet to temperature control substrate. (e) A fabricated microchip.
Detailed aptamer selection procedure
A schematic of the device setup can be seen in Figure 4. The device was interfaced with a syringe pump (New Era Pump Systems, Inc., Farmingdale, NY), a power supply (E3631, Agilent Technologies Inc., CA), and a multimeter (34420A, Agilent Technologies Inc., CA). During electrokinetic fluid handling platinum wires were inserted into the electrode ports and connected to the power supply. The multimeter was used to measure the resistance of the temperature sensor and ultimately interfaced with a computer which converted the resistance to a temperature using a calibration curve. The power supply was also interfaced to the computer which implemented a PID Labview (National Instruments, Austin, TX) algorithm for closed-loop temperature control in the chambers. The syringe pump was used for the buffer washes during affinity selection.

Figure 4. Experimental setup.
The device was first filled with tris-boric acid electrolyte buffer (TB buffer, 89 mM Tris, 89 mM boric acid, and 100 mM NaCl) and then target-functionalized magnetic beads (2 μL, 10 mg/mL) were introduced into the device using a pipette and an external magnet. A droplet merging technique was used where the liquid in the pipette tip was allowed to contact the fluid in the chip. Once this fluidic conduit was established an external magnet was used to pull the magnetic beads from the pipette tip into the chip. Library (1 μM, 100 μL in TB buffer) was then introduced into the selection chamber from the inlet (5 μL/min) and allowed to flow across the target-functionalized beads and ultimately to the outlet. An external magnet was held beneath the chamber to retain the beads in the selection chamber. The connecting microchannels provided sufficient fluidic resistance such that there was no observable flow to the amplification chamber or electrode port. Following a 30-minute incubation with the library, the chamber was washed with eight 5 minute long buffer washes at a flow rate of 20 μL/min. The wash buffer exiting the device during this washing process, containing weak binding oligonucleotides, was collected, amplified off-chip, and imaged with a gel electropherogram to verify the removal of weakly binding oligonucleotides from the device.
Amplification beads (2 μL, 10 mg/mL) were then introduced into the amplification chamber using a pipette and external magnet. The Labview PID algorithm was implemented to increase the selection chamber temperature to 55°C for 40 minutes thus inducing conformational changes in the oligonucleotides which disrupt the binding of the oligonucleotides to the target. Meanwhile a 40 V/cm electric field was applied for 40 minutes causing the thermally eluted oligonucleotides to migrate from the selection chamber to the amplification chamber wherein they bind to the amplification beads. The external magnet was kept beneath the chambers to retain the selection and amplification beads in their respective chambers. Following the migration process, PCR reagents were introduced into the amplification chamber using a pipette. A Labview algorithm was again implemented to thermally cycle the amplification chamber for 20 cycles of PCR (cycles of 5s at 95°C, 15s at 55°C, and 10s at 72°C). For characterization purposes, bead surfaces were imaged using an epifluorescence microscope (IX 71, Olympus, Center Valley, PA) equipped with a CCD (c8484, Hamamatsu, Boston, MA) before and after PCR, and after thermally induced ssDNA release.
At the conclusion of PCR, the external magnet was removed to allow the selection beads in the amplification chamber to be removed and replaced with fresh selection beads with a pipette. With fresh selection beads in the selection chamber, the amplification chamber was heated to 95°C inducing dehybridization of the single-stranded oligonucleotide from its complement and the electric field (40 V/cm) was again applied in reverse polarity to transfer the amplified oligonucleotides from the amplification chamber to the selection chamber. Once the oligonucleotides had travelled to the selection chamber, the selection chamber was again washed with eight 5 minute buffer washes at a flow rate of 20 μL/min. Following the wash the chamber was heated to 55°C using the integrated heater and temperature sensor and the eluted oligonucleotides were collected and analyzed for their affinity toward the target.
Aptamer characterization procedure
The enriched aptamer pool collected following completion of SELEX was investigated for its affinity using a fluorescence binding assay.1 The collected pool was amplified off-chip using a fluorescent forward primer and diluted to a final concentration of 100 nM. The 100 nM fluorescently tagged oligonucleotides were then incubated in triplicate 100 μL volumes with 20 μL (10 mg/mL) of washed target-functionalized beads. After incubation for 30 minutes, the beads were washed three times (100 μL each wash) and the bound oligonucleotides were eluted by heating to 95°C. The relative amount of eluted oligonucleotides was measured with a Wallac EnVision Multilabel Reader (PerkinElmer, Waltham, MA) fluorescent spectrometer. This was repeated with the library used to initiate the aptamer development process.
Results and Discussion
The device was first characterized to ensure the device could sufficiently perform the individual sub-processes (affinity selection, elution, electrokinetic transport, and PCR amplification). Then closed-loop affinity selection and amplification were performed on-chip consisting of two rounds of affinity selection and one round of PCR amplification. The device can readily be used for additional cycles of affinity selection and amplification when necessitated by the application.
Heater and temperature sensor characterization
The heater and temperature sensor were characterized by placing a fabricated device in an environmental chamber (Delta Designs 9023, Cohu, Poway, CA) and ramping the temperature from room temperature (∼23°C) to the maximum temperature used for PCR (95°C) in 10°C intervals. For every 10°C interval the resistance and temperature was recorded (Figure 5). By fitting this highly linear data a temperature coefficient of resistance was obtained using the following expression.
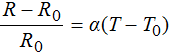

Figure 5. Characterization of the microchip temperature sensors. The linear relationship between temperature and resistor resistance for the (a) selection chamber and (b) amplification chamber resistors allows determination of a temperature coefficient of resistance.
In this expression R is the resistance for a given temperature, R0 is the resistance at room temperature, T is the temperature, T0 is the room temperature and alpha is the coefficient of resistance. A coefficient of resistance of 1.837 × 10−3°C−1 was obtained for the selection chamber temperature sensor and 1.902 × 10−3°C−1 for the amplification chamber temperature sensor.
Affinity selection
The affinity selection process must be able to provide an environment that allows potential aptamers in the initial starting library to bind to the target functionalized on the magnetic beads. The process must also be able to successfully partition binding oligonucleotides from weakly binding oligonucleotides. To verify successful affinity selection the initial starting library was introduced into the selection chamber which was preloaded with target-functionalized beads. Eight buffer washes were used to remove weakly binding oligonucleotides with effluents collected and analyzed. Finally the chamber temperature was increased to disrupt the binding between the binding oligonucleotides and the target molecules, and the chamber was washed using the same flow rate and volume of buffer that was used for the buffer washing. The effluent from this wash was collected and analyzed. All binding effluents were amplified off-chip by PCR (16 cycles) and imaged using gel electrophoresis (Figure 6). A decrease in band intensity in the gel electropherogram indicates fewer oligonucleotide molecules present. Thus, the decreasing band intensity from the first wash (W1) to the final wash (W8) indicates that in the initial washes many weakly binding oligonucleotides were removed from the selection chamber and as washing progressed few oligonucleotides remained that could be removed by washing alone. When the chamber was heated (55°C), represented by the elution (E) lane, a relatively high amount of oligonucleotides could be removed from the selection chamber. This suggests weakly binding oligonucleotides were removed by washing and upon heating the remaining strongly bound oligonucleotides could be eluted and collected or, if SELEX was to continue, transferred for PCR amplification. Therefore, the selection process was successful in portioning non-binding oligonucleotides, which were washed out of the chamber, from binding oligonucleotides, which could be eluted through a modest increase in temperature.

Figure 6. (a) Experiment schematic of the selection characterization. (b) Gel electropherogram and (c) measured band intensities of amplified eluents obtained during selection process with bar graph depicting intensities of lanes W1-E: washes 1, 2, 3, 4, 5, 6, 7, 8, and elution.
Electrokinetic transfer
Electrokinetic migration of oligomers was characterized by injecting fluorescently modified (fluorescein) oligomers into the selection chamber (100 nM) and applying an electric field (40 V/cm) with platinum wires from the selection chamber to the amplification chamber. While applying the electric field, an epifluorescent microscope (Eclipse 50i, Nikon, Tokyo, Japan) was focused on the amplification chamber and a micrograph of the chamber was acquired in one minute intervals with a charge-coupled device (Orca-ER, Hamamatsu Photonics, Hamamatsu, Japan). This process was repeated three times with a 40 V/cm electric field and three times without applying an electric field.
The fluorescent signal of the fluorescein-modified library is shown Figure 7. The error bars indicate the standard deviation of three repeated measurements, representing experimental errors associated with sources such as the imprecision in the volume of nucleotide solution dispensed into the inlet well and time variation of the electroknetic properties of the PDMS surfaces.45 When an electric field of 40 V/cm was applied, the intensity of fluorescence, representing the amount of oligonucleotides that had been transferred from the selection chamber to the amplification chamber, began increasing after 10 minutes and then saturated within approximately 37 minutes, In the absence of an electric field, the fluorescent intensity of the amplification chamber did not rise above background even for time durations longer than one hour (data not shown). Thus, to transfer oligonucleotides between functional chambers an electric field of 40 V/cm was applied for 40 minutes, which was considered sufficient for transferring oligonucleotides between the chambers.
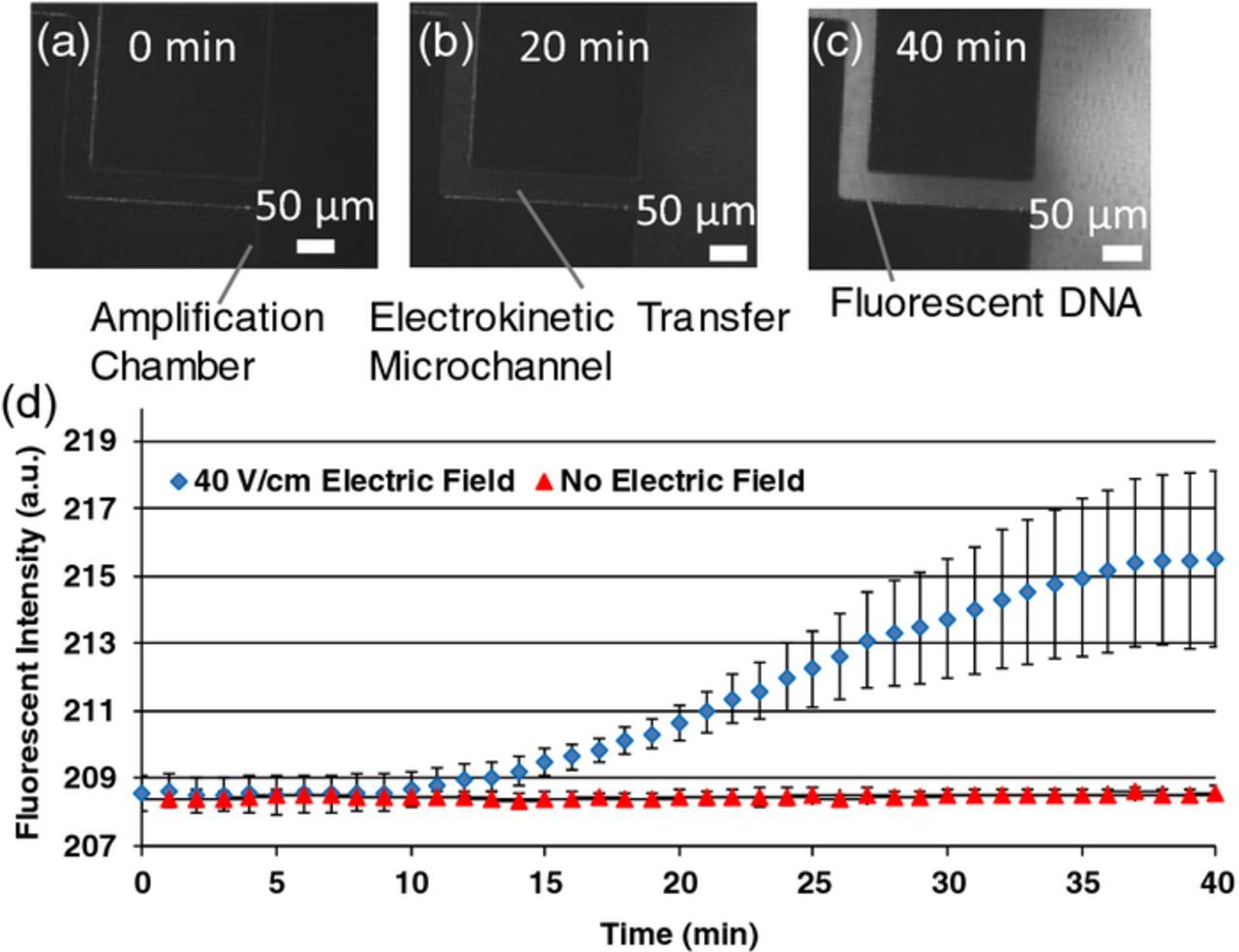
Figure 7. Fluorescent ssDNA was introduced to the selection chamber and fluorescent images were taken of the transfer microchannel at the amplification chamber after (a) 0 minutes, (b) 20 minutes, and (c) 40 minutes of applying a 40 V/cm electric field. (d) Time-course fluorescent intensity of selection chamber; error bars represent standard error; scale bars: 50 μm.
Considering a transfer channel 14.75 mm long, the oligonucleotide apparent velocity, vapp, was approximately 6.145 μm/s. Using this velocity along with the electric field strength, E, an apparent mobility electrokinetic, μapp, of the solution can be defined.

From this expression an apparent mobility of 1.536 × 10−8 m2/Vs is obtained which agrees with other PDMS/PDMS fabricated devices.46
Amplification
On-chip bead-based PCR was verified by injecting fluorescently modified oligonucleotides into the amplification chamber containing reverse-primer beads. PCR reagents, including a fluorescently modified forward primer, were then introduced and 20 cycles of PCR thermal cycling. Following thermal cycling the beads in the chamber were washed to remove non-incorporated fluorescent primers. The fluorescent intensity of the beads was measured before thermal cycling, after thermal cycling, and after thermal elution. As a control the process was repeated without including taq polymerase (Figure 8). The increase in brightness of the bead surfaces after the PCR thermal cycling suggests that the PCR process successfully amplified the oligomers on the bead surfaces, while the decrease intensity after thermal elution indicates that single-stranded oligonucleotides were thermally released from the bead surfaces. The fluorescent intensity of the beads used in the no-taq experiment did no increase after thermal cycling.
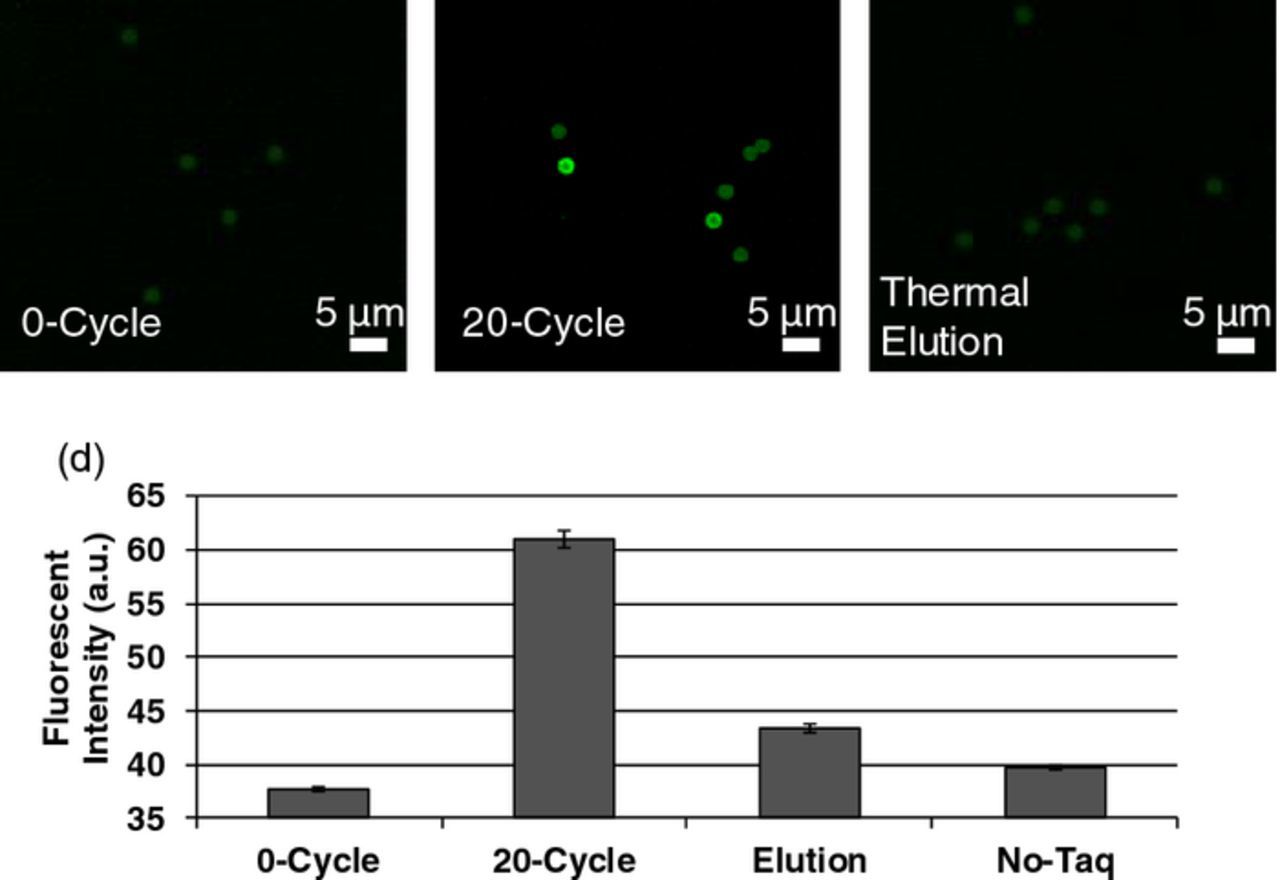
Figure 8. Fluorescent images of beads (A) before, (B) after 20 cycles of PCR, and (C) after 95°C thermally induced ssDNA elution. (D) Bar graph depicting fluorescent intensities. Error bars represent standard errors.
Closed-loop SELEX
The microchip performed microfluidic SELEX, which primarily involved integrated affinity selection and amplification, for isolation of aptamer candidates that bind to protein IgA1. To investigate the progress of the multi-round SELEX experiment, weakly bound ssDNA in each wash in the affinity selection procedure of each round, as well as the thermally eluted, strongly protein-binding oligonucleotides in the final round, were collected from the selection chamber, amplified and analyzed with gel electrophoresis (Figure 9). In the first wash of the first round (S1W1) there were a significant amount of oligonucleotides present but this decreased as the washing process progressed. In the first wash of the second round (S2W1) there were many more oligonucleotides present than the last wash from the first round (S1W8). This indicates successful transfer of the oligonucleotides to the amplification, PCR amplification and transfer back to the selection chamber for a second round of affinity selection. At the final wash of the second round (S2W8) there was not a detectable amount of oligonucleotides present; however, upon thermal elution (S2E) there were again detectable amounts of oligonucleotides. Thus, the affinity selection process in these rounds effectively removed weakly bound ssDNA as target-binding oligomers became more enriched. Moreover, high fluorescence intensity was observed in the thermal eluate (E) from the final round of SELEX, indicating that a significant amount of enriched protein-binding oligonucleotides (i.e., aptamer candidates) were generated from the SELEX process.
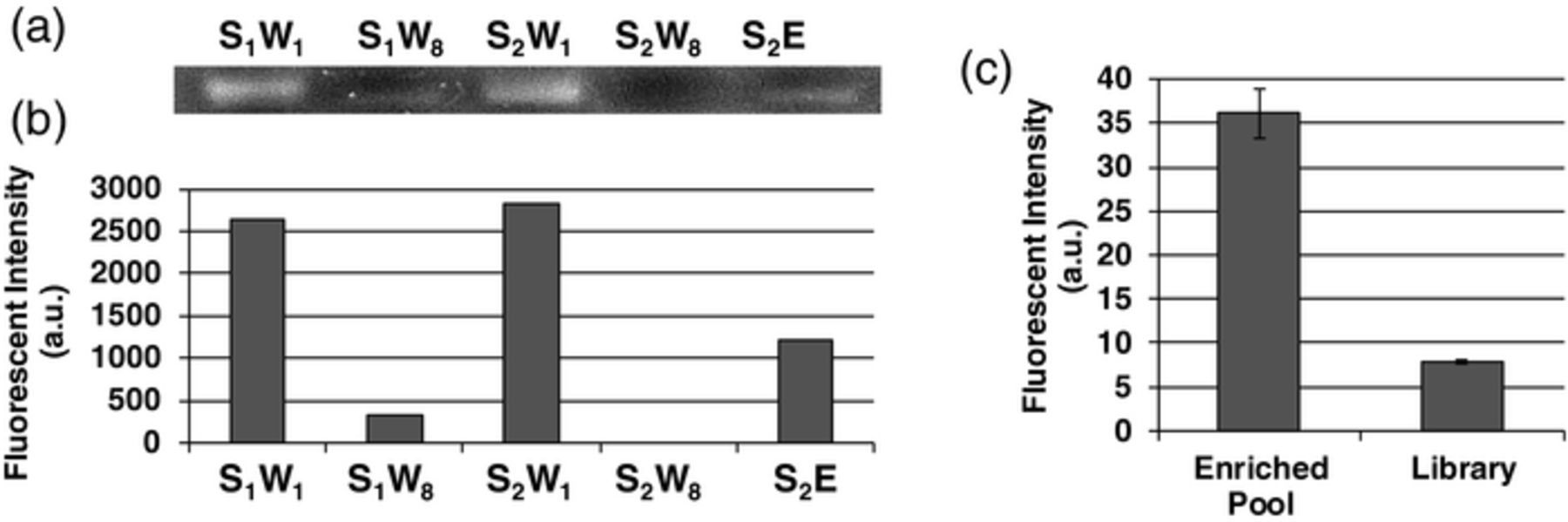
Figure 9. (a) Gel electropherogram of amplified eluents obtained during closed-loop selection and amplification. (b) Bar graph depicting intensities of lanes S1W1-S2E: S1W1: selection 1, wash 1; S1W10: selection 1, wash 10; S2W1: selection 2, wash 1; S2W10: selection 2, wash 10; S2E: oligomers released upon thermal elution. (c) Fluorescent intensity of fluorescently labeled enriched pool and library thermally eluted from target-functionalized beads after incubation and washing. Error bars represent standard error.
The oligonucleotides isolated from the microfluidic SELEX process were tested for their affinity toward IgA1 using a fluorescence binding assay. The background-subtracted average fluorescence intensity of the enriched aptamer candidate pool incubated with IgA was significantly higher than the fluorescence intensity of the library used to initiate the SELEX process. This indicates the enriched pool bound with considerably higher affinity and suggests that the mult-round SELEX process using the microchip was successful. Further enrichment of the candidate pool can be achieved with further rounds of affinity selection and amplification with the microchip.
Conclusions
We have presented an integrated, microfluidic approach to isolate aptamer against protein targets using a simplified electrokinetic transfer scheme. The approach involves bead-based reactions for electrokinetically coupled affinity selection and PCR amplification. The electrokinetic coupling allows highly integrated migration of oligonucleotides with reduced flow control equipment and without the need for complicated fabrication methods or gels, while also preventing undesirable diffusion or cross-flow effects. Thus, the approach is capable of integrating the iterative cycles of affinity selection and amplification onto a single chip without requiring offline processes that are commonly used in existing microfluidic aptamer selection devices. The approach was demonstrated by selecting an enriched pool of aptamers with affinity toward IgA. Oligonucleotides were affinity selected from a randomized library, electrokinetically transferred, PCR amplified, and transferred back for further affinity selection. The approach is readily capable of repeating these cycles of selection and amplification to obtain a pool of aptamers with sufficient affinity to a target of interest. The resulting enriched pool showed increased affinity toward the target when compared with the starting library. Future work involves implementing the device in clinical settings where rapidly generated affinity molecules are in demand with particular emphasis on precision medicine applications. Furthermore, the work will be extended to perform more cycles of the SELEX process on-chip to obtain aptamers with higher affinities.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (Award Nos. 8R21 GM104204, R01GM104960, 1R21CA199849, 2P41EB002033-19A1, 1R33CA196470, UL1 TR000040 and TL1 TR000082), and the Chinese Academy of Sciences SAFEA International Innovation Teams program.