
Abstract
In this work, we explicitly consider the evolution of the Standard Genetic Code (SGC) by assuming two evolutionary stages, to wit, the primeval RNY code and two intermediate codes in between. We used network theory and graph theory to measure the connectivity of each phenotypic graph. The connectivity values are compared to the values of the codes under different randomization scenarios. An error-correcting optimal code is one in which the algebraic connectivity is minimized. We show that the SGC is optimal in regard to its robustness and error-tolerance when compared to all random codes under different assumptions.
Similar content being viewed by others

Introduction
The standard genetic code (SGC) is almost universal. This feature supports the hypothesis of the existence of Last Common Ancestor Universal (LUCA) and it led to the proposal of Crick’s “Frozen accident hypothesis” (Crick 1968). This hypothesis states that the SGC does not change and was fixed by an accident. Arguing that changes in the SGC would be lethal or be strongly selected against, Crick stated the most accepted hypothesis for the evolution of the genetic code (Crick 1968). Different proposals for the origin and evolution of the genetic code have been made (Woese 1973; Rodin et al. 2011; Carter and Wolfenden 2015; Ribas de Pouplana and Schimmel 2001; Delarue 2007; Rodin and Rodin 2008; José et al. 2017; Wong 1988, 2005; Di Giulio 2013; Bandhu et al. 2013; Rouch 2014).
The SGC is a dictionary of three letter words based on the alphabet (A, U, C, G). The words are known as codons. Sixty-one codons codify for 20 amino acids and three codify the stop signals. Glycine is considered the first amino acid incorporated into the genetic code (Bernhardt and Patrick 2014; Tamura 2015). The SGC is commonly represented in a table (Table 1), with the codons written in the 5′ to 3′ direction, and it allows us to read off this code directly (Crick 1968). One property that immediately stands out from this representation is its degeneracy in the third base of some codons. This block structure accounts for the Crick’s wobble hypothesis (Crick 1958; Crick et al. 1976). The wobble hypothesis states that the third base is much less specific than the other two, and so, it is said to wobble and allow similar codons to codify for the same amino acid. The SGC is shown in Table 1, where the first and second bases of the codons arrange the amino acids in blocks, whereas the wobble property resides in the third base.
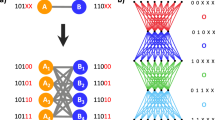
The RNY (purine-any base-pyrimidine) subcode is mostly considered as the primeval genetic code (Eigen and Schuster 1978; Eigen and Winkler-Oswatitsch 1981). It is composed by 16 codons and 8 amino acids, with two codons for each amino acid. Most of these amino acids were also found in Miller’s experiments and observations on abundant amino acids and in meteorites (Miller 1953; Miller et al. 1976). The SGC has been theoretically derived from the RNY code through a process of symmetry breakings (José et al. 2007, 2014) that were identified with two evolutionary pathways. The first path is a degenerate RNA code which can be translated in the 1st (RNY), 2nd (NYR), and the 3rd (YRN) reading frames, and the second path is obtained by transversions in the 1st (YNY) and 3rd (RNR) nucleotide bases of the 16 codons of the RNA subcode. The composition of both intermediate subcodes led straightforward to the SGC using the RNY as primeval code. The SGC was mathematically modeled in a hypercube of six dimensions (6D) where the vertices of the cube represent the codons and the edges join codons that differ in a single nucleotide (José et al. 2017). In the 6D–hypercube, the RNY code is represented by a 4-dimensional hypercube (Zamudio and José 2017) and the evolutionary steps are expansions to higher dimensions in order to finally reach the 6D–hypercube of the SGC. Phenotypic graph representations of amino acids based on the topology of the SGC hypercube has been developed (José et al. 2015). A phenotypic graph is one in which the vertices represent the 21 signals of the SGC and the edges join amino acids or the stop signal based on the adjacencies of the SGC hypercube (José et al. 2014, 2015; Zamudio and José 2017).
The current block structure of the genetic code has also been analyzed to understand the processes involved in the assignment of amino acids to codons (Woese et al. 1966; Caporaso et al. 2005). The robustness of the SGC and its capacity to tolerate errors in translation by misreading of the codons have been analysed (Alff-Steinberger 1969; Ardell and Sella 2002). This feature was analysed with random codes (Novozhilov et al. 2007). Stochastic simulations showed that the SGC is not optimal for error-minimization but when other biological properties were taken into account it was found to be suboptimal in a fitness landscape (Wong 1980; Haig and Hurst 1991; Freeland et al. 2000; Novozhilov et al. 2007).
In this work, we usher in both network and graph theory as a novel approach to examine the evolution of the genetic code. We analyze the connectivity properties of the phenotypic graphs that are extracted from the genetic codes that arise from two evolutionary pathways assuming RNY as a primeval code. The measure known as algebraic connectivity reflects the general connectivity of a network (de Abreu 2007; Newman 2010). As this measure gets higher, a network is more connected. The application of this measure to the phenotypic graphs will determine the connectivity of the 21 signals of the SGC and reflect the associations of the codons in the 6D–hypercube model. These connectivity values are compared to the values of the codes under different randomization scenarios. The codon – amino acid associations are fixed in every stage of the evolution of the genetic code before proceeding to the next evolutionary step.
We show that the SGC is indeed optimal by analyzing the algebraic connectivity of the phenotypic graphs of the SGC, the extended codes (intermediate evolutionary steps), and the RNY code (primeval code).
Methods
Graph theory is a branch of discrete mathematics devoted to the analysis of graphs. A graph is an object composed by vertices and edges. The vertices represent objects of any kind and the edges reflect some relation between those objects (Newman 2010). Several measures have been developed to describe the connectivity of a graph. Most of them assign to each vertex a centrality or connectivity score (Newman 2010). The algebraic connectivity is a connectivity measure for a set of vertices or for the whole graph. This measure is denoted by α(G), where G stands for a graph. It is given by the second smallest eigenvalue of the Laplacian matrix of G (de Abreu 2007; Newman 2010) and represents a parameter to measure the connectivity of a graph. The adjacency matrix is important in graph theory because it captures the entire structure of a network and whose matrix properties are used to characterize the properties of edges and vertices. There is another matrix, the Laplacian matrix closely related to the adjacency matrix that can also tell us much about the network structure. In fact, the Laplacian matrix of a graph G is given by, the adjacency matrix of G minus the identity matrix. The Laplacian is a symmetric matrix, and so has real eigenvalues. All the eigenvalues of the Laplacian are also non-negative. While the eigenvalues of the Laplacian cannot be negative, they can be zero, and in fact the Laplacian always has at least one zero eigenvalue. Since there are no negative eigenvalues, this is the lowest of the eigenvalues of the Laplacian. By convention, the eigenvalues are numbered in ascending order: λ 1 ≤ λ 2 ≤ … ≤ λ n . So we always have λ 1 = 0. The second eigenvalue of the graph Laplacian λ 2 is non-zero if and only if the network is connected. The algebraic connectivity is widely used in algorithms aimed to expand graphs, and the eigenvector associated to the algebraic connectivity, known as Fielder vector, is used for combinatorial optimization problems (de Abreu 2007).
Inhere, the evolutionary pathways of the SGC with a RNY subcode as its ancestor is considered. The genetic codes from each stage of the evolution path are subject to different degrees of randomization and set in the 6D–dimensional representation of the SGC, in which the vertices of a 6D–hypercube represent the codons, and the edges join codons that differ by a single nucleotide (José et al. 2017). Then, its phenotypic graphs were calculated. The phenotypic graph represents the phenotypic expression of the codon hypercube; the vertices represent the 20 amino acids and the stop signal. In the phenotypic graph, two signals (amino acid or stop signal) of the genetic code are joined if there exist two codons for those signals that are adjacent in the 6D–model (José et al. 2014, 2015; Zamudio and José 2017). A lower algebraic connectivity in the phenotypic graphs reflects a general absence of edges joining the amino acids in the phenotypic graph. In turn, the latter reflects that the codons codifying for the same amino acid are joined, and therefore, they are similar, instead of being randomly scattered across the hypercube. An error-correcting optimal code is one in which the algebraic connectivity is minimized. If the amino acids were randomly associated to the codons, the block structure of the genetic code vanishes and the resulting phenotypic graph would present a high algebraic connectivity.
Starting from the RNY subcode, two evolutionary pathways have been proposed (José et al. 2009, 2014). One path comprehends an extension of the RNY subcode by means of frame-shift reading mistranslations; this generates the codes NYR and YRN in addition to the original RNY. This path is known as the Extended code type I (Ex1). The second path is reached by transversions in the first and the third base of the RNY subcode; this code comprises the codons of the form RNR and YNY. The second path is known as Extended code type II (Ex2). By complementing both Extended codes, the rest of the codons are generated and the SGC is completed to 64 codons.
For the RNY subcode, random codes in which the amino acids were randomly permutated and assigned to the 16 RNY codons were calculated. RNYp denotes these random codes. Three levels of randomization were calculated for each of the extended codes. Ex1s and Ex2s denote the codes that resulted from a random permutation of the amino acids and stop signal of the SGC and then the extended codes were extracted. Ex1p and Ex2p denote the codes that arise form a restricted permutation of the signals present in each of the extended codes. Finally, Ex1d and Ex2d denote the extended codes with a random degeneracy of the signals present on each code. For the complete genetic code, three distinct levels of randomization were also considered. Codes with random degeneracy of the 21 signals are denoted by SGCr. Codes with the amino acids and the stop signal randomly permutated are SGCp. The third level is denoted by SGCrd in which all the properties of the wobble and the degeneracy are preserved; the wobbling is present in the third base. The SGCrd codes represents codes that only permutate blocks that maintain the third wobbling base of the SGC. For each random code, 5000 permutations were calculated and the algebraic connectivity for each phenotypic graph obtained by each random code case was computed.
Results
For all the randomized codes, the range of values or intervals between the minimum and maximum of the algebraic connectivity is presented in Table 2. The algebraic connectivity of the RNY, Ex1, Ex2 and SGC are also included. For all the random control codes of the Extended codes, none of them presented a minimum of algebraic connectivity that falls below the algebraic connectivity of both Ex1 and Ex2. There were only 3 codes out of the 5000 random codes of the RNY code, that presented the same connectivity as the actual RNY whose connectivity is equal to 2. These three codes (RNYp) differ from the actual RNY by a misplacement of Glycine in the triplets GGC and GGU (Table 3).
In the Ex2s randomization, 4 out of the 5000 permutations presented a lower connectivity than the actual Extended code type II. These permutations displayed alterations in the RNY code. Recall that the random controls of the SGC were as follows: pure random degeneracy (SGCr), fixed degeneracy without wobbling (SGCp), and fixing wobbling but shuffling the codon-amino acids assignments (SGCrd). Note in Table 2 that the maximum values of the connectivity of the random controls of the SGC, decrease as more restrictions to the controls are added, reflecting that codons codifying for the same amino acid tend to be closer together in the 6D–hypercube. The hexacodonic amino acids Serine, Leucine and Arginine, present two codons that are less similar than the other four in which the third base wobble, especially Serine. In other words, in hexacodonic amino acids, if the 6 codons are more similar among them (to keep the second base invariant), then the connectivity of the phenotypic graph will decrease. Some random codes of the SGC present a lower connectivity than the current SGC. The SGCp random control showed one code and the SGCrd presented 1825 codes with lower connectivity. All of them presented modifications in the RNY subcode and in the Extended codes. Details of the codes with lower algebraic connectivity are presented in Appendix.
Discussion
We show that the SGC is indeed optimal when its evolutionary stages from the primeval RNY code, and the extended codes are considered. This result was achieved by calculating the connectivity properties of the phenotypic graphs of each code. The different degrees of randomization applied to the SGC and the Extended codes allowed measuring the effect of different properties of the genetic code to assign the same amino acid to similar codons. The RNY subcode and the Extended codes delineate concrete stages to analyze the robustness and error correction properties of the genetic code, in contrast to previous approaches which ignored the evolution of the SGC (Haig and Hurst 1991; Novozhilov et al. 2007). The randomized codes did show that more optimal codes exist as found in other works (Wong 1980; Haig and Hurst 1991; Freeland et al. 2000; Novozhilov et al. 2007). However, the codes that resulted with a lower connectivity than the present SGC exhibited modifications on the initial stages of the evolutionary pathway, i.e. the amino acids associated to codons of the Extended codes or the RNY subcode are different from the ones currently present in those codes. In a general perspective the codes with lower connectivity would be more resistant to errors, yet their occurrence would require major codon swaps to achieve those codes and would not be evolutionary optimal as proposed by Di Giulio (1989). Codon swaps for the reassignment of amino acids to triplets has been proposed as one of the principal mechanisms for the evolution of the genetic code (Szathmáry 1991). In between the evolutionary stages of the SGC, derived by the symmetry-breaking model, codon swaps must have presumably occurred in order to produce the present assignments of those codons. Once any of the two Extended codes were reached it was fixed and no amino acid reassignments further occurred; the same fixation is extended to the core RNY subcode. The scaling properties of the distance series of each codon of the RNY, Extended codes, and the SGC show critical scale invariance and this property is a universal vestige in genomes of Eubacteria and Archea (José et al. 2009).
The 6D–model of the genetic code (José et al. 2007) has been shown to be equivalent to the Rodin-Ohno model (Rodin and Ohno 1995) of the genetic code (José et al. 2017). It shows symmetries relative to the Woese’s polar requirement scales of amino acids (Woese et al. 1966), and to the partition of the code derived by the class of aminoacyl-tRNA synthetases (aaRSs) associated to the amino acids (José et al. 2017). When restricted to the RNY subcode, its phenotypic graph has been coupled with the division of aaRS in order to derive the biological properties that uniquely identify the present SGC (Zamudio and José 2017). The regularity of this core code is reflected in its phenotypic graphs (José et al. 2015) when a SGC model in lower dimensions is considered (José et al. 2012).
de Farías et al. (2014) proposed an origin for the coding system based on the co-evolution of tRNAs and aaRs, and further driven by changes in the second base of the anticodon that affected its hydropathy. Other mechanisms driving the evolution of the genetic code include the polarity of amino acids (Di Giulio 1989), and the configuration of peptides formed by the genetic code (Alff-Steinberger 1969). All these pressures fixed and froze the SGC at different stages when the codons of the evolutionary paths were assigned to the amino acids that constitute a genetic code more robust and with high error-tolerance capacity, leading ultimately to the completion SGC.
References
Alff-Steinberger C (1969) The genetic code and error transmission. Proc Natl Acad Sci U S A 64:584–591. https://doi.org/10.1073/pnas.64.2.584
Ardell DH, Sella G (2002) No accident: genetic codes freeze in error-correcting patterns of the standard genetic code. Philos Trans R Soc Lond Ser B Biol Sci 357:1625–1642. https://doi.org/10.1098/rstb.2002.1071
Bandhu AV, Aggarwal N, Sengupta S (2013) Revisiting the physico-chemical hypothesis of code origin: an analysis based on code-sequence coevolution in a finite population. Orig Life Evol Biosph 43:465–489. https://doi.org/10.1007/s11084-014-9353-x
Bernhardt HS, Patrick WM (2014) Genetic code evolution started with the incorporation of glycine, followed by other small hydrophilic amino acids. J Mol Evol 78:307–309. https://doi.org/10.1007/s00239-014-9627-y
Caporaso JG, Yarus M, Knight R (2005) Error minimization and coding triplet/binding site associations are independent features of the canonical genetic code. J Mol Evol 61:597–607. https://doi.org/10.1007/s00239-004-0314-2
Carter CW, Wolfenden R (2015) tRNA acceptor stem and anticodon bases form independent codes related to protein folding. Proc Natl Acad Sci 112:7489–7494. https://doi.org/10.1073/pnas.1507569112
Crick FHC (1958) On protein synthesis. Symp Soc Exp Biol 12:138–166
Crick FHC (1968) The origin of the genetic code. J Mol Biol 38:367–379. https://doi.org/10.1016/0022-2836(68)90392-6
Crick FHC, Brenner S, Klug A, Pieczenik G (1976) A speculation on the origin of protein synthesis. Orig Life 7:389–397. https://doi.org/10.1007/BF00927934
de Abreu NMM (2007) Old and new results on algebraic connectivity of graphs. Linear Algebra Appl 423:53–73. https://doi.org/10.1016/j.laa.2006.08.017
de Farías ST, do Rêgo TG, José MV (2014) Evolution of tranfer RNA and the origin of the translation system. Front Genet 5:303
Delarue M (2007) An asymmetric underlying rule in the assignment of codons: possible clue to a quick early evolution of the genetic code via successive binary choices. RNA 13:161–169. https://doi.org/10.1261/rna.257607
Di Giulio M (1989) The extension reached by the minimization of the polarity distances during the evolution of the genetic code. J Mol Evol 29:288–293. https://doi.org/10.1007/BF02103616
Di Giulio M (2013) The origin of the genetic code: matter of metabolism or physicochemical determinism? J Mol Evol 77:131–133. https://doi.org/10.1007/s00239-013-9593-9
Eigen M, Schuster P (1978) The hypercycle - a principle of natural self-organization part b: the abstract hypercycle. Naturwissenschaften 65:7–41. https://doi.org/10.1007/BF00420631
Eigen M, Winkler-Oswatitsch R (1981) Transfer-RNA, an early gene? Naturwissenschaften 68:282–292. https://doi.org/10.1007/BF01047470
Freeland SJ, Knight RD, Landweber LF, Hurst LD (2000) Early fixation of an optimal genetic code. Mol Biol Evol 17:511–518. https://doi.org/10.1093/oxfordjournals.molbev.a026331
Haig D, Hurst LD (1991) A quantitative measure of error minimization in the genetic-code. J Mol Evol 33:412–417. https://doi.org/10.1007/bf02103132
José MV, Morgado ER, Govezensky T (2007) An extended RNA code and its relationship to the standard genetic code: an algebraic and geometrical approach. Bull Math Biol 69:215–243. https://doi.org/10.1007/s11538-006-9119-3
José MV, Govezensky T, García JA, Bobadilla JR (2009) On the evolution of the standard genetic code: vestiges of critical scale invariance from the RNA world in current prokaryote genomes. PLoS One. https://doi.org/10.1371/journal.pone.0004340
José MV, Morgado ER, Sanchez R, Govezensky T (2012) The 24 possible algebraic representations of the standard genetic code in six or in three dimensions. Adv Stud Biol 4:119–152
José MV, Morgado ER, Guimarães RC et al (2014) Three-dimensional algebraic models of the tRNA code and 12 graphs for representing the amino acids. Life (Basel, Switzerland) 4:341–373. https://doi.org/10.3390/life4030341
José MV, Zamudio GS, Palacios-Pérez M et al (2015) Symmetrical and thermodynamic properties of phenotypic graphs of amino acids encoded by the primeval RNY code. Orig Life Evol Biosph 45:77–83. https://doi.org/10.1007/s11084-015-9427-4
José MV, Zamudio GS, Morgado ER (2017) A unified model of the standard genetic code. R Soc Open Sci 4:160908. https://doi.org/10.1098/rsos.160908
Miller SL (1953) A production of amino acids under possible primitive earth conditions. Science (80- ) 117:528–529. https://doi.org/10.1126/science.117.3046.528
Miller SL, Urey HC, Oró J (1976) Origin of organic compounds on the primitive earth and in meteorites. J Mol Evol 9:59–72. https://doi.org/10.1007/BF01796123
Newman MEJ (2010) Networks : an introduction. Oxford University Press, Oxford
Novozhilov AS, Wolf YI, Koonin EV (2007) Evolution of the genetic code: partial optimization of a random code for robustness to translation error in a rugged fitness landscape. Biol Direct 2:24. https://doi.org/10.1186/1745-6150-2-24
Ribas de Pouplana L, Schimmel P (2001) Aminoacyl-tRNA synthetases: Potential markers of genetic code development. Trends Biochem Sci 26:591–596
Rodin SN, Ohno S (1995) Two types of aminoacyl-trna synthetases could be originally encoded by complementary strands of the same nucleic ACID. Orig Life Evol Biosph 25:565–589. https://doi.org/10.1007/BF01582025
Rodin SN, Rodin AS (2008) On the origin of the genetic code: signatures of its primordial complementarity in tRNAs and aminoacyl-tRNA synthetases. Heredity (Edinb) 100:341–355. https://doi.org/10.1038/sj.hdy.6801086
Rodin AS, Szathmáry E, Rodin SN (2011) On origin of genetic code and tRNA before translation. Biol Direct 6:14. https://doi.org/10.1186/1745-6150-6-14
Rouch DA (2014) Evolution of the first genetic cells and the universal genetic code: a hypothesis based on macromolecular coevolution of RNA and proteins. J Theor Biol 357:220–244. https://doi.org/10.1016/j.jtbi.2014.06.003
Szathmáry E (1991) Codon swapping as a possible evolutionary mechanism. J Mol Evol 32:178–182. https://doi.org/10.1007/BF02515390
Tamura K (2015) Beyond the frozen accident: glycine assignment in the genetic code. J Mol Evol 81:69–71
Woese CR (1973) Evolution of the genetic code. Naturwissenschaften 60:447–459. https://doi.org/10.1007/BF00592854
Woese CR, Dugre DH, Saxinger WC, Dugre SA (1966) The molecular basis for the genetic code. Proc Natl Acad Sci U S A 55:966–974. https://doi.org/10.1073/pnas.55.4.966
Wong JT (1980) Role of minimization of chemical distances between amino acids in the evolution of the genetic code. Proc Natl Acad Sci U S A 77:1083–1086
Wong JT (1988) Evolution of the genetic code. Microbiol Sci 5:174–181
Wong JT-F (2005) Coevolution theory of the genetic code at age thirty. BioEssays 27:416–425. https://doi.org/10.1002/bies.20208
Zamudio GS, José MV (2017) On the uniqueness of the standard genetic code. Life (Basel, Switzerland) 7:7. https://doi.org/10.3390/life7010007.
Acknowledgments
Gabriel S. Zamudio is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and a fellowship recipient from Consejo Nacional de Ciencia y Tecnología (CONACYT) (number: 737920). M.V.J. was financially supported by PAPIIT-IN224015; UNAM; México. We thank Juan R. Bobadilla for the technical computer support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(XLSX 616 kb)
Rights and permissions
About this article

Cite this article
Zamudio, G.S., José, M.V. Phenotypic Graphs and Evolution Unfold the Standard Genetic Code as the Optimal. Orig Life Evol Biosph 48, 83–91 (2018). https://doi.org/10.1007/s11084-017-9552-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-017-9552-3