
Abstract
Cerebral cavernous malformation is a neurovascular abnormality that can cause seizures, focal neurological deficits and intracerebral hemorrhage. Familial forms of this condition are characterized by de novo formation of multiple lesions and are autosomal-dominantly inherited via CCM1/KRIT1, CCM2/MGC4607 and CCM3/PDCD10 mutations. We identified three truncating mutations in KRIT1 from three Japanese families with CCMs: a novel frameshift mutation, a known frameshift mutation and a known splice-site mutation that had not been previously analyzed for aberrant splicing.
Similar content being viewed by others
Cerebral cavernous malformation (CCM) is a vascular malformation in the central nervous system characterized by clusters of enlarged capillary cavities without intervening brain parenchyma.1 Clinical manifestations can vary from asymptomatic or mild symptoms, such as headache, to more severe symptoms, such as seizures, focal neurological deficits and hemorrhage.2 The prevalence of CCM in the general population has been reported to range from 0.39 to 0.53% according to large series of autopsies and magnetic resonance imaging (MRI) studies.3,4 The familial form of CCM is particularly common in Hispanic-American patients with CCMs (~50%); this form is distinguishable from the sporadic form owing to the multiplicity and de novo formation of CCMs.5,6 Three autosomal-dominantly inherited loci, i.e., CCM1 (7q21.2, MIM*604214), CCM2 (7p13, MIM*607929) and CCM3 (3q26.1, MIM*609118), have been identified to date; within these respective loci, the Krev interaction trapped-1 (KRIT1), malcavernin (MGC4607) and programmed cell death 10 (PDCD10) genes act as causative genes of familial CCMs.5 In the present study, we analyzed three Japanese families with segregating CCMs; two of these families included patients presenting de novo formation of CCM lesions and multiple cerebral and spinal lesions confirmed by MRI follow-up over a decade.
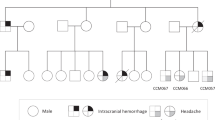
The first pedigree (pedigree A) harbored at least 10 affected members according to anamnestic data (Figure 1a). The proband (II:1) was a 63-year-old man. At 53 years of age, he was incidentally diagnosed with multiple CCMs on cerebral MRI (Figure 1c). Although he was asymptomatic during the follow-up period of 10 years, the most recent MRI showed a large number of de novo lesions in the brain compared with that at the initial diagnosis (Figure 1d). Because of his characteristic MRI findings and family history, his four children (III:1–4) underwent cerebral MRI despite the absence of clinical symptoms. As expected, their MRIs also showed multiple CCMs, with the exception of the daughter (III:3). The three affected sons (III:1, 2 and 4) were all asymptomatic during the follow-up period of 9 years; however, two of his sons (III:2 and 4) developed de novo CCMs in follow-up MRIs (data not shown). Detailed clinical data of the other affected relatives in this pedigree are not available.

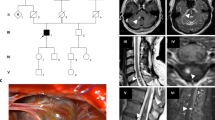
Clinical manifestations in the individuals in this study. (a, b) Genealogical trees of pedigrees A and B. Black-filled symbols: clearly affected individuals; symbol enclosing a vertical bar: asymptomatic carrier; crossed-out symbols: deceased individuals; arrows: probands. DNA sequencing was carried out in individuals denoted with an asterisk. (c–k) Magnetic resonance imaging of patients. T2-weighted axial brain images of II:1 in pedigree A at diagnosis (c) and those after 10 years (d). T2*-weighted axial brain images of III:5 in pedigree B at diagnosis (e) and those after 7 years (f). T2-weighted axial brain images of his brother (III:6) at onset (g) and those at 4 years after surgery (h). White arrows: de novo cerebral cavernous malformations (CCMs); open arrow: a hypothalamic CCM before surgery. The T2-weighted sagittal spinal image of II:3 in pedigree B (i). The T2-weighted sagittal spinal image (j) and T2*-weighted axial brain images (k) of the proband of pedigree C before surgery.
The second pedigree (pedigree B) harbored at least six affected members according to anamnestic data (Figure 1b). The proband (II:5) was a 60-year-old man, who was also incidentally diagnosed with multiple CCMs on cerebral MRI at 50 years of age. However, he refused follow-up imaging studies because he remained asymptomatic after diagnosis. His 32-year-old son (III:5) presented with transient left hemiparesis associated with a CCM in the right side of the central area at 25 years of age (Figure 1e). Follow-up MRI at 7 years after onset showed multiple de novo lesions in the brain (Figure 1f). The other son of the proband (III:6) presented with consciousness disturbance associated with a hypothalamic CCM at 18 years of age (Figure 1g). His consciousness was recovered after surgical removal of the lesion; however, he suffered from permanent hypothalamic–pituitary dysfunction. At 22 years of age, he developed cerebellar hemorrhage from a de novo CCM (Figure 1h). Although he appeared to have recovered, he died suddenly of pulmonary embolism at 3 months after a second surgical removal. The proband’s 63-year-old brother (II:3) presented with numbness in the bilateral lower extremities at 62 years of age. Spinal MRI showed a spinal cavernous malformation (SCM) at the level of Th 12 (Figure 1i). Cerebral MRI has not yet been performed for this patient. The proband’s daughter (III:7) and the other family members were also not screened with cerebral MRI because they were clinically asymptomatic.
The third pedigree (pedigree C) consisted of a parent–offspring pair of patients. The proband was a 76-year-old father, whose daughter suffered intracerebral hemorrhage due to CCM; he presented with right shoulder pain at 73 years of age, and his symptoms progressed to sensory and motor disturbance of the right upper extremity, with eventual deterioration to tetraparesis at 7 months after onset. His MRI showed a cervical SCM with perifocal edema, indicating recent bleeding (Figure 1j) and multiple CCMs (Figure 1k). He remained severely disabled, even after the SCM was resected.
The Ethical Committee of Tokyo Women’s Medical University approved the study protocols. After obtaining written informed consent, genomic DNA samples were obtained from 12 participants: the three affected members (II:1, III:1 and III:2) in pedigree A (Figure 1a); the three affected members (II:3, II:5 and III:5), three presumed intrafamilial controls (I:2, II:1 and II:6) and two relatives of unknown disease status (II:7 and III:7) in pedigree B (Figure 1b), and the proband in pedigree C (Table 1). PCR-based direct Sanger sequencing was performed in all coding exons and their exon/intron junctions for the KRIT1 (NM_194456.1), MGC4607 (NM_031443.3) and PDCD10 (NM_007217.3) genes. From this analysis, three pathogenic mutations in KRIT1 were detected from each of the three families, reflecting high allelic heterogeneity in patients with familial CCMs except for Hispanic-American patients harboring the founder p.Gln248Ter mutation (c.742C>T, rs267607203) in this gene.1,7 There were no pathogenic mutations in MGC4607 or PDCD10.
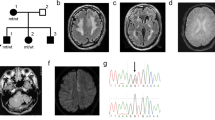
The KRIT1 mutation in pedigree A was a heterozygous transition at a splice donor site of intron 10 (c.845+1G>A; Figure 2a). This mutation was shared among the three affected participants and was absent from the 1000 Genomes Project database (http://www.1000genomes.org/);8 NHLBI GO Exome Sequencing Project (Exome Variant Server, http://evs.gs.washington.edu/EVS/); NCBI dbSNP144 (http://www.ncbi.nlm.nih.gov/snp); and Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/).9 To test the impact of this mutation on splicing, total RNA samples were obtained from peripheral blood leukocytes of the proband (II:1) and an unrelated healthy volunteer without CCMs confirmed by cerebral MRI (PAXgene Blood RNA Kit; Qiagen, Valencia, CA, USA). Complementary DNA (cDNA) was synthesized using a Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannheim, Germany). Reverse transcription PCR covering a 799-bp cDNA sequence from exon 8 to exon 13 was performed using the following primers: 5′- GTAGTGAATCCAGTACTCATTTTGC-3′ and 5′- GCAGCTTCTTCCCAGTTGTT-3′, which showed altered splicing patterns for the mutant allele (Figure 2b). Sequencing of the subcloned PCR products (Mighty TA-cloning Kit; TaKaRa, Shiga, Japan) revealed three types of splicing variants. Although the second smallest fragment in the gel electrophoresis was a known splicing isoform lacking exon 10 (NM_001013406.1), the smallest fragment contained two aberrantly spliced variants having similar nucleotide sizes. One skipped exons 10 and 11, and the other skipped 131 bp in the end of exons 9 and 10, generating the immediate premature termination sequences p.Asp245TrpfsTer4 and p.Gln301AsnfsTer5, respectively (Figure 2c).

Results of genetic analysis. (a) DNA sequence chromatogram of the pedigree A proband (II:1), showing the heterozygous c.845+1G>A mutation of KRIT1. (b) Agarose gel electrophoresis of reverse transcriptase PCR (RT-PCR) products covering exons 8–13 of KRIT1, showing altered splicing patterns of the c.845+1G>A allele. (c) Sequencing of clones obtained from the RT-PCR amplicons revealed three types of splicing alterations. (d, e) DNA sequence chromatograms of frameshift KRIT1 mutations detected in pedigrees B and C.
In pedigrees B and C, we identified two heterozygous frameshift mutations, i.e., c.1362_1363delTC (Figure 2d) and c.1153delA (Figure 2e), respectively. These mutations also resulted in the generation of premature terminations (p.Gln455ArgfsTer24 and p.Thr385GlnfsTer9) and were not listed in the above-mentioned four public databases. In pedigree B, the c.1362_1363delTC mutation was found in the three affected patients, but was not identified in the three intrafamilial controls. III:7 was an asymptomatic carrier who had not undergone cerebral MRI (Figure 1b).
These mutations were screened against the CCM Mutation Database (http://www.angiomaalliance.org/pages.aspx?content=345&id=289), NCBI ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and PubMed (http://www.ncbi.nlm.nih.gov/pubmed). The c.845+1G>A mutation10 in pedigree A and the c.1362_1363delTC mutation11,12 in pedigree B have been reported previously; however, until the current study, no functional studies had been performed to validate the impaired splicing of the c.845+1G>A mutation. The c.1153delA mutation in pedigree C had not been previously reported.
Patients with CCM often remain asymptomatic,2,5 whereas SCM tends to be more clinically progressive,12 consistent with the clinical manifestations observed in our patients (Table 1). Early SCM resection has been reported to be associated with good outcomes,13,14 although our patient with lumbar SCM (II:3, pedigree B) showed no neurological aggravation during conservative therapy. Therefore, both cerebral and spinal MRI should be considered for patients with multiple CCMs and their potentially affected relatives in order to achieve early diagnosis of subclinical SCM. Conversely, cerebral MRI should be also considered for patients with SCM and their relatives because CCMs occur in 17–42% of patients with SCM, and 12–57% of patients with SCM have a family history of cavernous malformation.13,15
KRIT1, encoded by the KRIT1 gene, interacts with the other two CCM proteins encoded by MGC4607 and PDCD10 to form a heterotrimeric CCM complex.16 This complex regulates various signaling pathways represented by Rho/ROCK, whose inhibitor fasudil has been shown to reduce CCM lesions in Krit1-knockdown mice.17,18 In a recent study, endothelial gain of mitogen-activated protein kinase kinase kinase 3 (MEKK3)-Krüppel-like factor (KLF) 2/4 signaling was shown to be a key biological process during early CCM formation due to disruption of the CCM complex. Endothelial-specific inactivation of Mekk3 and its target genes Klf2 and Klf4 prevents CCM formation in neonatal mice with endothelial-specific deletion of Krit1.19 For future therapies targeting such promising biological pathways, genetic testing and improved knowledge of pathogenic mutations in CCM genes will become increasingly important.
References
References
Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 1999; 23: 189–193.
Washington CW, McCoy KE, Zipfel GJ . Update on the natural history of cavernous malformations and factors predicting aggressive clinical presentation. Neurosurg Focus 2010; 29: E7.
Del Curling O Jr, Kelly DL Jr, Elster AD, Craven TE . An analysis of the natural history of cavernous angiomas. J Neurosurg 1991; 75: 702–708.
Robinson JR, Awad IA, Little JR . Natural history of the cavernous angioma. J Neurosurg 1991; 75: 709–714.
Labauge P, Denier C, Bergametti F, Tournier-Lasserve E . Genetics of cavernous angiomas. Lancet Neurol 2007; 6: 237–244.
Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT et al. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med 1988; 319: 343–347.
Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet 1999; 8: 2325–2333.
1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65.
Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet 2016; 61: 547–553.
Spiegler S, Najm J, Liu J, Gkalympoudis S, Schröder W, Borck G et al. High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one-third of probands are minors. Mol. Genet Genomic Med 2014; 2: 176–185.
Mondéjar R, Solano F, Rubio R, Delgado M, Pérez-Sempere A, González-Meneses A et al. Mutation prevalence of cerebral cavernous malformation genes in Spanish patients. PLoS ONE 2014; 9: e86286.
Riant F, Cecillon M, Saugier-Veber P, Tournier-Lasserve E . CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics 2013; 14: 133–141.
Badhiwala JH, Farrokhyar F, Alhazzani W, Yarascavitch B, Aref M, Algird A et al. Surgical outcomes and natural history of intramedullary spinal cord cavernous malformations: a single-center series and meta-analysis of individual patient data. J Neurosurg Spine 2014; 21: 662–676.
Zevgaridis D, Medele RJ, Hamburger C, Steiger HJ, Reulen HJ . Cavernous haemangiomas of the spinal cord. A review of 117 cases. Acta Neurochir 1999; 141: 237–245.
Cohen-Gadol AA, Jacob JT, Edwards DA, Krauss WE . Coexistence of intracranial and spinal cavernous malformations: a study of prevalence and natural history. J Neurosurg 2006; 104: 376–381.
Fisher OS, Boggon TJ . Signaling pathways and the cerebral cavernous malformations proteins: lessons from structural biology. Cell Mol Life Sci 2014; 71: 1881–1892.
McDonald DA, Shi C, Shenkar R, Stockton RA, Liu F, Ginsberg MH et al. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke 2012; 43: 571–514.
Stockton RA, Shenkar R, Awad IA, Ginsberg MH . Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med 2010; 207: 881–896.
Zhou Z, Tang AT, Wong WY, Bamezai S, Goddard LM, Shenkar R et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 2016; 532: 122–126.
Data Citations
Akagawa, Hiroyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.849
Akagawa, Hiroyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.852
Akagawa, Hiroyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.855
Acknowledgements
We thank the patients and their families for making this study possible. This study was supported by Funds for the Development of Human Resources in Science and Technology to the Program to Disseminate Tenure Tracking System (HA) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT, FY2011).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hirota, K., Akagawa, H., Kikuchi, A. et al. KRIT1 mutations in three Japanese pedigrees with hereditary cavernous malformation. Hum Genome Var 3, 16032 (2016). https://doi.org/10.1038/hgv.2016.32
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.32
