Physiologically-Based Pharmacokinetic/Pharmacodynamic Model of MBQ-167 to Predict Tumor Growth Inhibition in Mice

 ,
,  ,
,  , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal Protocol
2.3. Tumor Pharmacokinetics
2.4. Tumor Sample Preparation
2.5. Instrumentation
2.6. Her2+/Triple Negative Tumor Growth Study
2.7. Whole Body Fluorescence Image Analysis
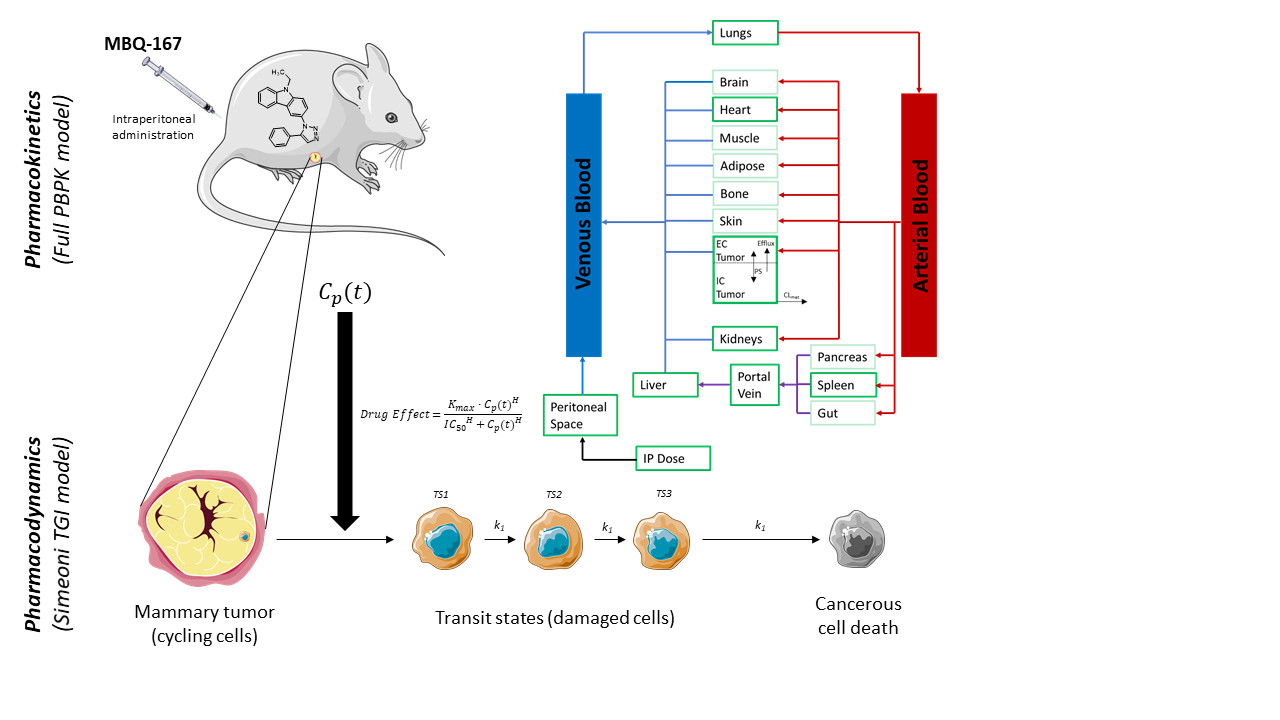
2.8. Physiologically Based Pharmacokinetic Model
2.8.1. Modelling Strategy
2.8.2. Physicochemical Properties and Plasma Binding
2.8.3. Absorption
2.8.4. Distribution
2.8.5. Elimination
2.8.6. Tumor Disposition
2.8.7. Population
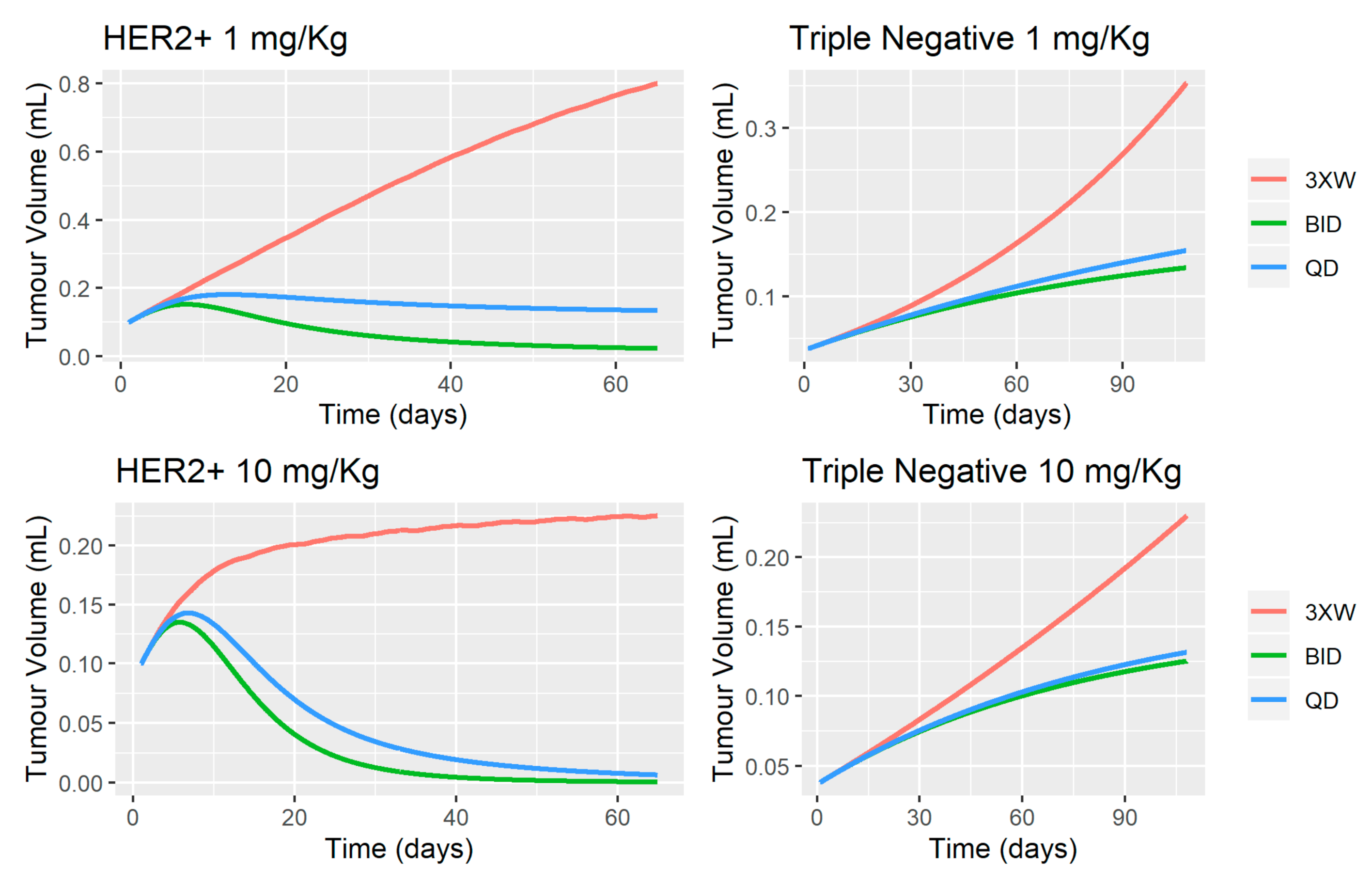
2.8.8. Simulation Trials
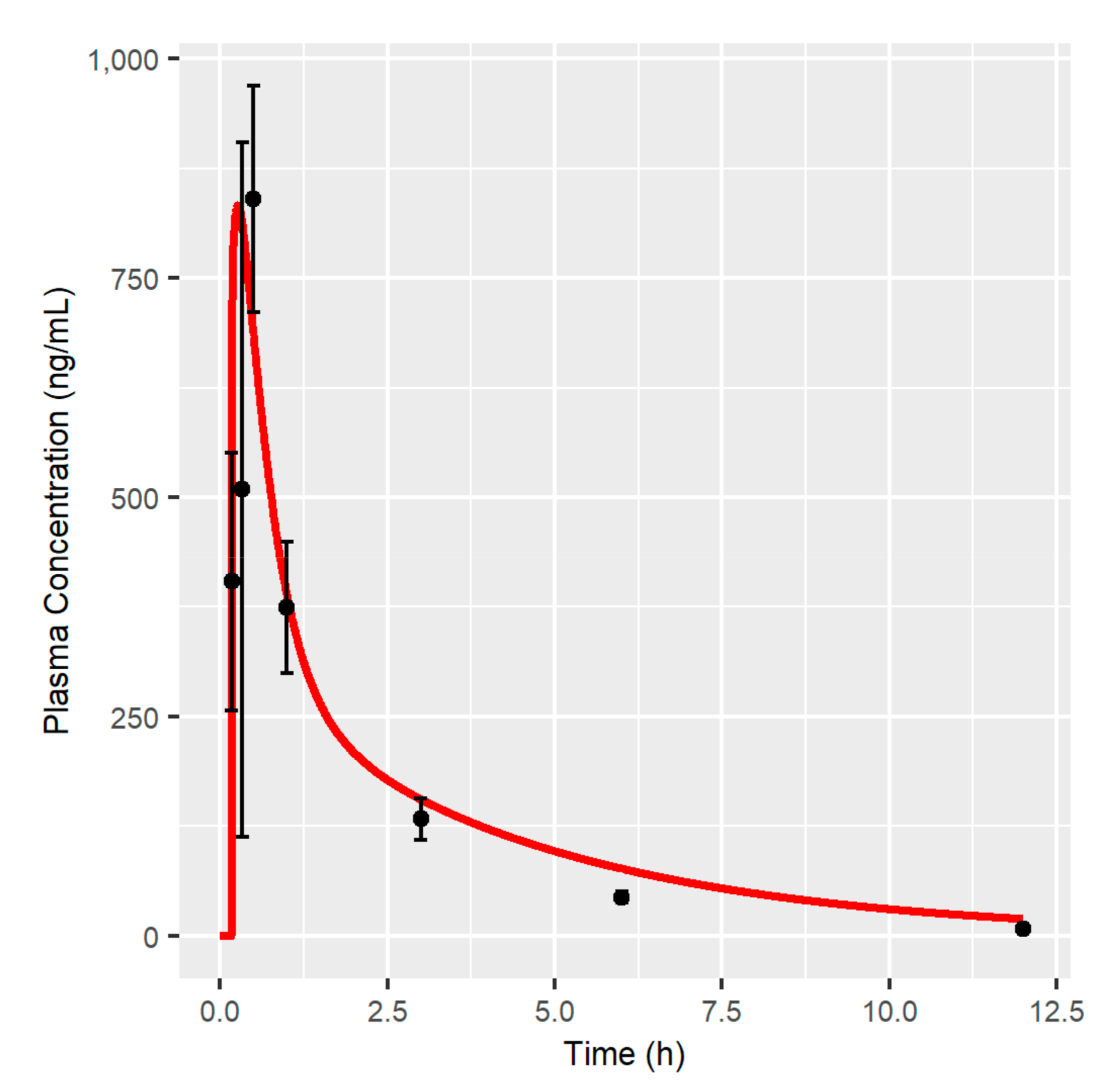
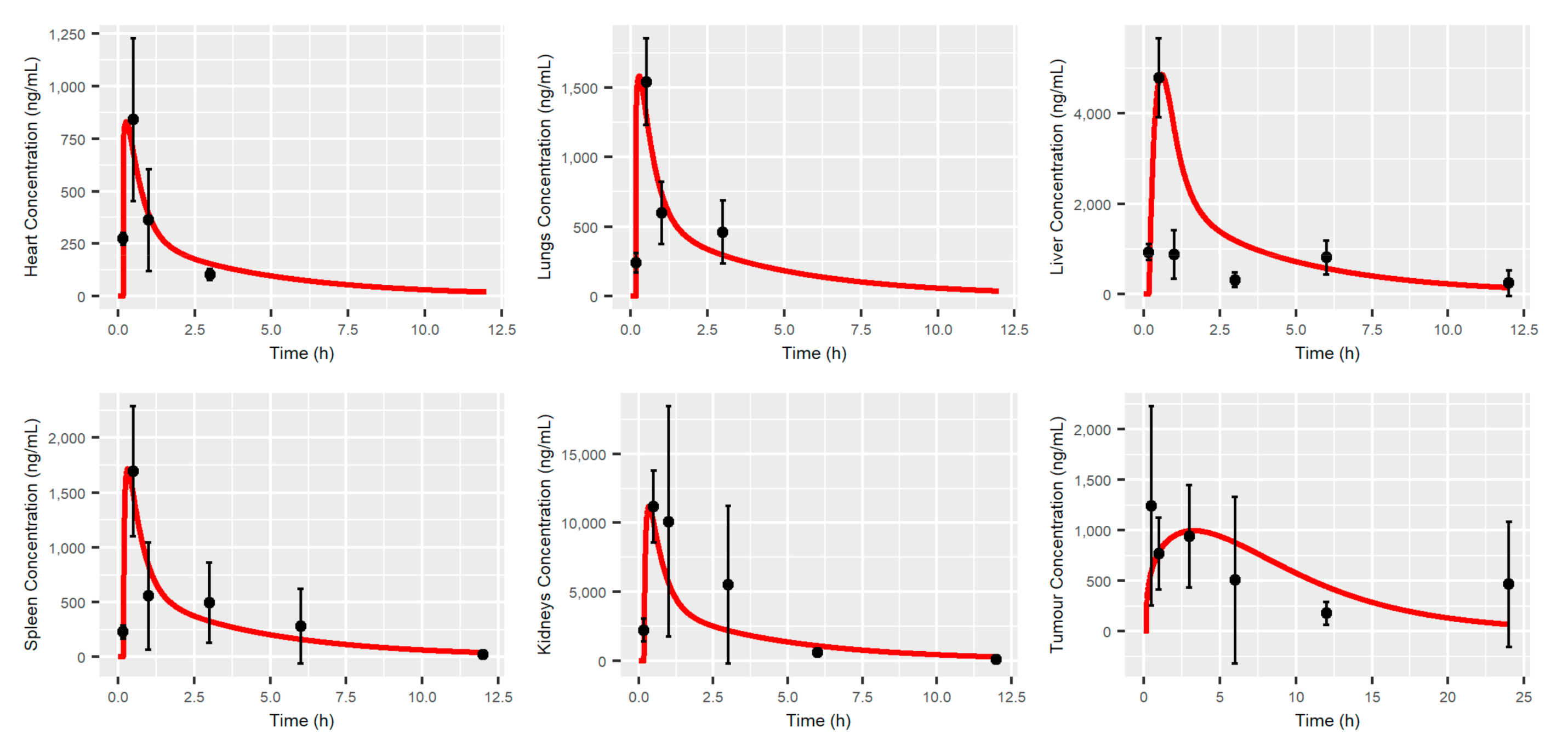
2.9. MBQ-167 PBPK Model Verification
2.10. MBQ-167 Tumor Growth Inhibition Model Development
3. Results
3.1. MBQ-167 PBPK Model
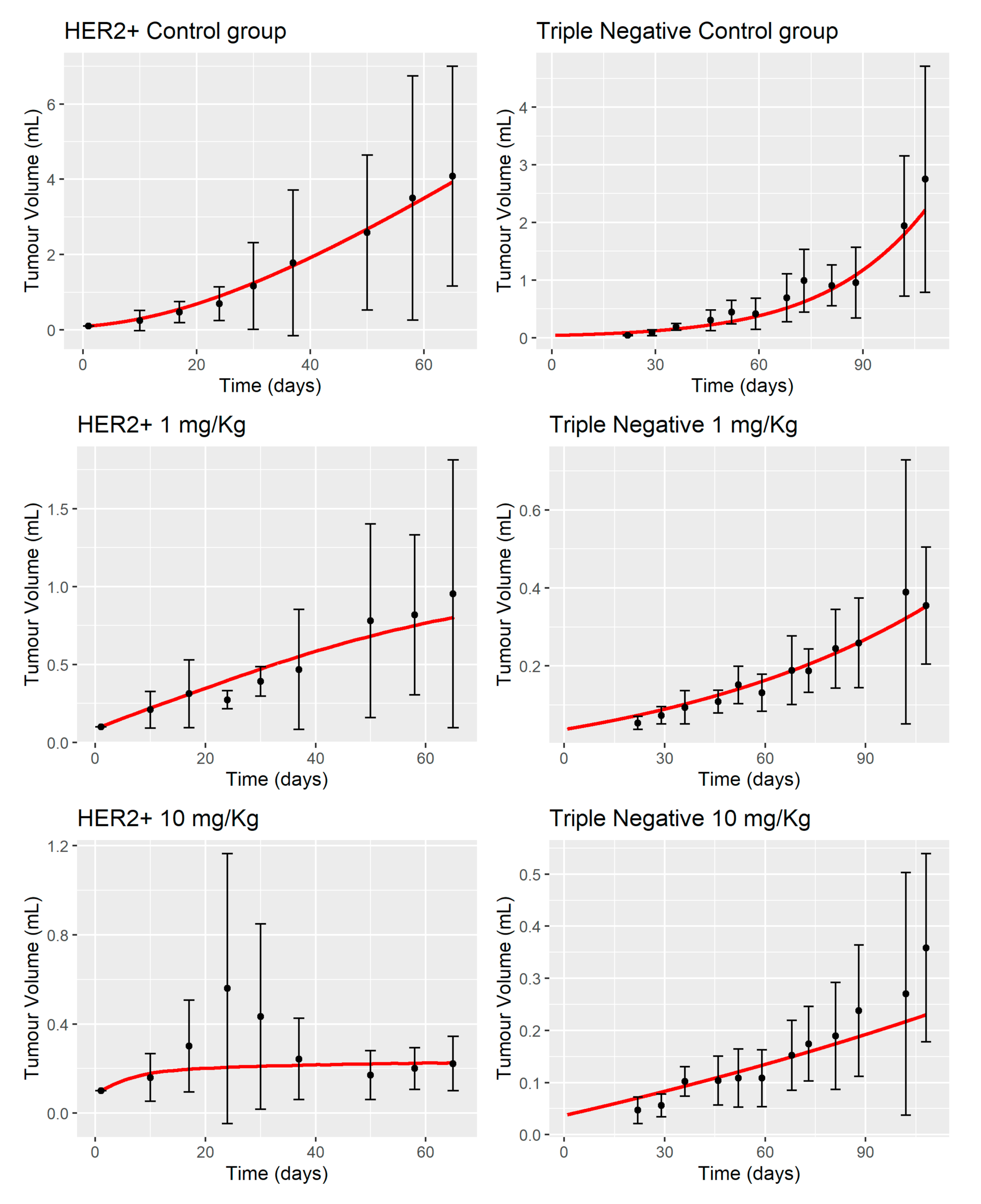
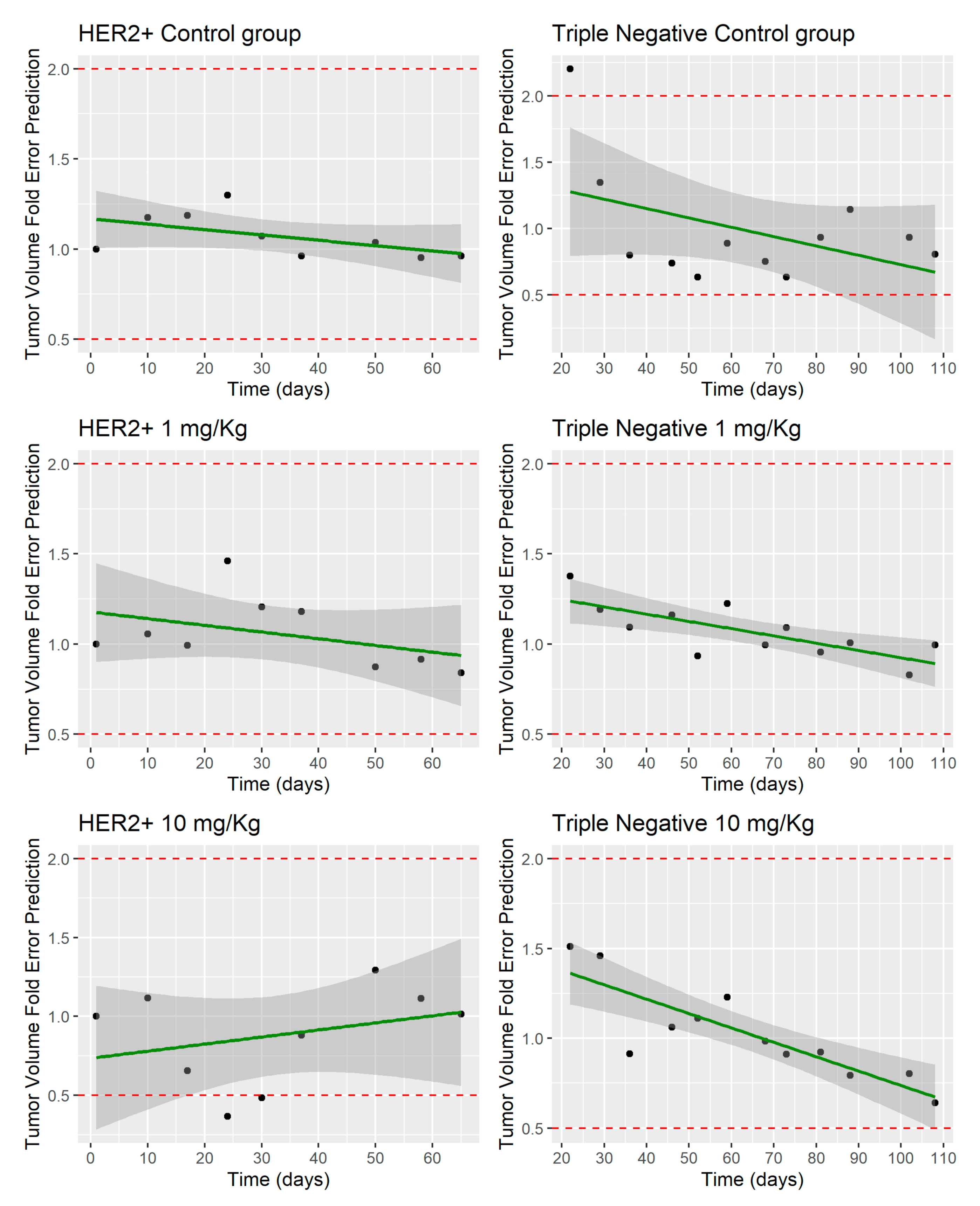
3.2. MBQ-167 Tumor Growth Inhibition Model
4. Discussion
4.1. MBQ-167 PBPK Model
4.2. MBQ-167 Tumor Growth Inhibition Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 95%CI | 95% confidence intervals |
| B/P | blood-to-plasma ratio |
| CLR | renal clearance |
| fu | unbound fraction |
| IC50 | concentration related to half of the maximal inhibitory concentration |
| IP | intraperitoneal |
| LSA | local sensitivity analysis |
| PBPK | Physiologically based pharmacokinetic |
| PBPK-PD | Physiologically based pharmacokinetic/pharmacodynamic |
| TGI | tumor growth inhibition |
| Vss | volume of distribution at steady state |
References
- Mansinho, A.; Boni, V.; Miguel, M.; Calvo, E. New designs in early clinical drug development. Ann. Oncol. 2019, 30, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Pearson, A.D.J. How can attrition rates be reduced in cancer drug discovery? Expert Opin. Drug Discov. 2013, 8, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sinha, V.; Forgue, S.T.; Callies, S.; Ni, L.; Peck, R.; Allerheiligen, S.R.B. Model-Based Drug Development: The Road to Quantitative Pharmacology. J. Pharmacokinet. Pharmacodyn. 2006, 33, 369–393. [Google Scholar] [CrossRef] [PubMed]
- Clewell, H.J.; Andersen, M.E. Physiologically-Based Pharmacokinetic Modeling and Bioactivation of Xenobiotics. Toxicol. Ind. Health 1994, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.N.; Simpson, P.M.; McCarver, D.G. Age-Dependent Human Hepatic Carboxylesterase 1 (CES1) and Carboxylesterase 2 (CES2) Postnatal Ontogeny. Drug Metab. Dispos. 2016, 44, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.; Clewell, H.J. Addressing Early Life Sensitivity Using Physiologically Based Pharmacokinetic Modeling and in Vitro to in Vivo Extrapolation. Toxicol. Res. 2016, 32, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.; Dutreix, C.; Rebello, S.; Ouatas, T.; Wang, L.; Chun, D.Y.; Einolf, H.J.; He, H. Simultaneous Physiologically Based Pharmacokinetic (PBPK) Modeling of Parent and Active Metabolites to Investigate Complex CYP3A4 Drug-Drug Interaction Potential: A Case Example of Midostaurin. Drug Metab. Dispos. 2018, 46, 109–121. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation_en.pdf (accessed on 9 August 2020).
- US Food and Drug Administration. Physiologically Based Pharmacokinetic Analyses—Format and Content Guidance for Industry. Available online: https://www.fda.gov/media/101469/download (accessed on 9 August 2020).
- Simeoni, M.; Magni, P.; Cammia, C.; De Nicolao, G.; Croci, V.; Pesenti, E.; Germani, M.; Poggesi, I.; Rocchetti, M. Predictive Pharmacokinetic-Pharmacodynamic Modeling of Tumor Growth Kinetics in Xenograft Models after Administration of Anticancer Agents. Cancer Res. 2004, 64, 1094–1101. [Google Scholar] [CrossRef] [Green Version]
- Parra-Guillen, Z.P.; Berraondo, P.; Ribba, B.; Trocóniz, I.F. Modeling Tumor Response after Combined Administration of Different Immune-Stimulatory Agents. J. Pharmacol. Exp. Ther. 2013, 346, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Tate, S.C.; Burke, T.F.; Hartman, D.; Kulanthaivel, P.; Beckmann, R.P.; Cronier, D.M. Optimising the combination dosing strategy of abemaciclib and vemurafenib in BRAF-mutated melanoma xenograft tumours. Br. J. Cancer 2016, 114, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.C.; Cai, S.; Ajamie, R.T.; Burke, T.; Beckmann, R.P.; Chan, E.M.; De Dios, A.; Wishart, G.N.; Gelbert, L.M.; Cronier, D.M. Semi-Mechanistic Pharmacokinetic/Pharmacodynamic Modeling of the Antitumor Activity of LY2835219, a New Cyclin-Dependent Kinase 4/6 Inhibitor, in Mice Bearing Human Tumor Xenografts. Clin. Cancer Res. 2014, 20, 3763–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terranova, N.; Germani, M.; Del Bene, F.; Magni, P. A predictive pharmacokinetic–pharmacodynamic model of tumor growth kinetics in xenograft mice after administration of anticancer agents given in combination. Cancer Chemother. Pharmacol. 2013, 72, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhu, X.; Han, M.; Hao, F.; Lu, W.; Zhou, T. Mechanistic Pharmacokinetic/Pharmacodynamic Model of Sunitinib and Dopamine in MCF-7/Adr Xenografts: Linking Cellular Heterogeneity to Tumour Burden. AAPS J. 2020, 22, 45. [Google Scholar] [CrossRef]
- Yang, L.; Yong, L.; Zhu, X.; Feng, Y.; Fu, Y.; Kong, D.; Lu, W.; Zhou, T. Disease progression model of 4T1 metastatic breast cancer. J. Pharmacokinet. Pharmacodyn. 2020, 47, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Lestini, G.; Mentré, F.; Magni, P. Optimal Design for Informative Protocols in Xenograft Tumor Growth Inhibition Experiments in Mice. AAPS J. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Hernandez-O’Farrill, E.; Borrero-Garcia, L.D.; Forestier-Roman, I.; Gerena, Y.; Blanco, M.; Rivera-Robles, M.J.; Rodriguez-Medina, J.R.; Cubano, L.A.; et al. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther. 2017, 16, 805–818. [Google Scholar] [CrossRef] [Green Version]
- Kazanietz, M.G.; Caloca, M.J. The Rac GTPase in Cancer: From Old Concepts to New Paradigms. Cancer Res. 2017, 77, 5445–5451. [Google Scholar] [CrossRef] [Green Version]
- Stengel, K.; Zheng, Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [Green Version]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef]
- De, P.; Aske, J.C.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Robles, M.J.; Medina-Velazquez, J.; Asencio-Torres, G.M.; Gonzalez-Crespo, S.; Rymond, B.C.; Rodriguez-Medina, J.; Dharmawardhane, S. Targeting Cdc42 with the anticancer compound MBQ-167 inhibits cell polarity and growth in the budding yeast S. cerevisiae. Small GTPases 2018, 1–11. [Google Scholar] [CrossRef]
- Al-abd, A.M.; Aljehani, Z.K.; Gazzaz, R.W.; Fakhri, S.H.; Jabbad, A.H.; Alahdal, A.M.; Torchilin, V.P. Pharmacokinetic strategies to improve drug penetration and entrapment within solid tumors. J. Control. Release 2015. [Google Scholar] [CrossRef]
- Del Mar Maldonado, M.; Rosado-González, G.; Bloom, J.; Duconge, J.; Ruiz-Calderón, J.F.; Hernández-O’Farrill, E.; Vlaar, C.; Rodríguez-Orengo, J.F.; Dharmawardhane, S. Pharmacokinetics of the Rac/Cdc42 Inhibitor MBQ-167 in Mice by Supercritical Fluid Chromatography-Tandem Mass Spectrometry. ACS Omega 2019, 4, 17981–17989. [Google Scholar] [CrossRef]
- The National Research Council; Commitee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011; Volume 46, ISBN 9780309154000. [Google Scholar]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac inhibitor EHop-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Creative Commons Attribution 3.0 Unported License Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 14 October 2020).
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Corujo-Carro, F.; Duconge, J.; Hernandez-O’Farrill, E.; Vlaar, C.; Rodriguez-Orengo, J.F.; Cubano, L.; Dharmawardhane, S. Pharmacokinetics of Rac inhibitor EHop-016 in mice by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2015, 981, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Li, J.; Rong, L.F.; Lü, X.W.; Huang, Y.; Xu, S.Y. Pharmacokinetics and tissue distribution of 5-fluorouracil encapsulated by galactosylceramide liposomes in mice. Acta Pharmacol. Sin. 2005, 26, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Center for Drug Evaluation and Research. Pharmacology Review(s). Application Number: 22-059. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022059s000_ClinPharmR.pdf (accessed on 13 October 2020).
- Hampsch, R.A.; Shee, K.; Bates, D.; Lewis, L.D.; Désiré, L.; Leblond, B.; Demidenko, E.; Stefan, K.; Huang, Y.H.; Miller, T.W. Therapeutic sensitivity to Rac GTPase inhibition requires consequential suppression of mTORC1, AKT, and MEK signaling in breast cancer. Oncotarget 2017, 8, 21806–21817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, B.K.; Boyer, A.; Ciccolini, J.; Barlesi, F.; Wang, K.; Benzekry, S.; Mochel, J.P. Optimal Scheduling of Bevacizumab and Pemetrexed/Cisplatin Dosing in Non-Small Cell Lung Cancer. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Kobuchi, S.; Shimizu, R.; Ito, Y. Semi-Mechanism-Based Pharmacokinetic-Toxicodynamic Model of Oxaliplatin-Induced Acute and Chronic Neuropathy. Pharmaceutics 2020, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Wang, S.; Ren, Y.; Li, J.; Guo, T.; Lu, W.; Zhou, T. Antitumor effect of axitinib combined with dopamine and PK-PD modeling in the treatment of human breast cancer xenograft. Acta Pharmacol. Sin. 2019, 40, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chen, R.; Yao, Q.-Y.; Liu, S.-J.; Tian, X.-Y.; Hao, C.-Y.; Lu, W.; Zhou, T.-Y. Time-dependent pharmacokinetics of dexamethasone and its efficacy in human breast cancer xenograft mice: A semi-mechanism-based pharmacokinetic/pharmacodynamic model. Acta Pharmacol. Sin. 2018, 39, 472–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, J.L.; Cabanillas, F.; Rivera, N.; Tirado-Gomez, M.; Hallman, D.; Pardo, W.I.; Bruno, M. Results of Upfront Therapy for Marginal Zone Lymphoma. Clin. Lymphoma. Myeloma Leuk. 2017, 17, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.W.T.; Holt, S.V.; Logie, A.; Payne, K.; Woods, K.; Wilkinson, R.W.; Davies, B.R.; Guichard, S.M. A pharmacokinetic-pharmacodynamic model predicting tumour growth inhibition after intermittent administration with the mTOR kinase inhibitor AZD8055. Br. J. Pharmacol. 2017, 174, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Wang, S.; Zhu, X.; Xue, J.; Li, J.; Wang, L.; Li, J.; Lu, W.; Zhou, T. Pharmacokinetic-Pharmacodynamic Modeling of the Anti-Tumor Effect of Sunitinib Combined with Dopamine in the Human Non-Small Cell Lung Cancer Xenograft. Pharm. Res. 2017, 34, 408–418. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Section | Parameter (Units) | Value | Source/Reference/Comments |
|---|---|---|---|
| Molecular Weight (g/mol) | 338.414 | ||
| logPo:w | 4.944 | ||
| Compound Type | Monoprotic Base | ||
| pKa | 0.27 | ||
| B/P | 1.8 | Optimized | |
| fu plasma | 0.02 | Estimated | |
| Distribution | Full PBPK | ||
| Vss (L/kg) | 20.21 | Simcyp predicted (Method 2) | |
| Kpt Heart | 1 | Optimized | |
| Kpt Kidney | 13.94 | Optimized | |
| Kpt Liver | 14.66 | Optimized | |
| Kpt Lung | 1.9 | Optimized | |
| Kpt Spleen | 2.1 | Optimized | |
| Kpt Scalar | 0.29 | Optimized | |
| Tumor | Permeability Limited Model | ||
| PS (mL/min/mL tumor volume) | 1.2 | Optimized | |
| Efflux transporter (mL/min mL tumor) | 7 | Estimated | |
| Elimination | |||
| Hep intrinsic CL (mL/min/106 cells) | 79 | In vitro determined | |
| fu_inc | 0.07 | Optimized | |
| Typical Renal CL (mL/min) | 0.3 | Assumed based on maximum glomerular filtration rate for mice [26]. | |
| Tumor CL (mL/min/mL tumor volume) | 2.2 | Optimized | |
| Administration Route | Other site | ||
| Dose (mg) | 0.2 | ||
| Condition | Fed | ||
| Input site | Venous Blood | Optimized | |
| Input model | First order | Optimized | |
| Lag time (h) | 0.17 | Optimized | |
| fa | 1 | ||
| ka (1/h) | 3 | Optimized | |
| Parameter\Cell Line | HER2+ | Triple Negative |
|---|---|---|
| System Related Parameters | ||
| Tumor growth model | Simeoni | Simeoni |
| Initial tumor volume (mL) | 0.1 a | 0.0384 b |
| λ0 (day−1) | 0.2 c | 0.0393 b |
| λ1 (g/day) | 0.12 c | 0.5457 b |
| Ψ | 0.7 c | 0.9985 b |
| Number of transit compartments | 3 | 4 |
| Drug Related Parameters | ||
| Response Model | Emax | Emax |
| Drug input | Total plasma concentration | Total plasma concentration |
| k1 (day−1) | 0.39 c | 0.0007 b |
| IC50 (μM) | 0.0187 b | 0.0001 b |
| Kmax (day−1) | 0.3683 b | 0.0533 c |
| H | 0.5 c | 0.5 c |
| Tissue | AUCobs | AUCpred | AUCpred/AUCobs | Cmax,obs | Cmax,pred | Cmax,pred/Cmax,obs |
|---|---|---|---|---|---|---|
| Plasma | 1417.2 | 1549.1 | 1.09 | 839.9 | 833.31 | 0.99 |
| Lung | 1887.6 | 1838.2 | 0.97 | 1540.9 | 1583.3 | 1.03 |
| Liver | 8388.5 | 11,444.3 | 1.36 | 4793.7 | 4865.6 | 1.01 |
| Spleen | 3983.9 | 3243.5 | 0.81 | 1693.4 | 1718.4 | 1.01 |
| Kidneys | 34279 | 21,527.9 | 0.63 | 11,160.4 | 11,231.7 | 1.00 |
| Heart | 949.2 | 965.4 | 1.02 | 840.8 | 831.2 | 0.99 |
| Tumor | 10,286.8 | 11,492.8 | 1.12 | 1243.6 | 997.0 | 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reig-López, J.; Maldonado, M.d.M.; Merino-Sanjuan, M.; Cruz-Collazo, A.M.; Ruiz-Calderón, J.F.; Mangas-Sanjuán, V.; Dharmawardhane, S.; Duconge, J. Physiologically-Based Pharmacokinetic/Pharmacodynamic Model of MBQ-167 to Predict Tumor Growth Inhibition in Mice. Pharmaceutics 2020, 12, 975. https://doi.org/10.3390/pharmaceutics12100975
Reig-López J, Maldonado MdM, Merino-Sanjuan M, Cruz-Collazo AM, Ruiz-Calderón JF, Mangas-Sanjuán V, Dharmawardhane S, Duconge J. Physiologically-Based Pharmacokinetic/Pharmacodynamic Model of MBQ-167 to Predict Tumor Growth Inhibition in Mice. Pharmaceutics. 2020; 12(10):975. https://doi.org/10.3390/pharmaceutics12100975
Chicago/Turabian StyleReig-López, Javier, María del Mar Maldonado, Matilde Merino-Sanjuan, Ailed M. Cruz-Collazo, Jean F. Ruiz-Calderón, Victor Mangas-Sanjuán, Suranganie Dharmawardhane, and Jorge Duconge. 2020. "Physiologically-Based Pharmacokinetic/Pharmacodynamic Model of MBQ-167 to Predict Tumor Growth Inhibition in Mice" Pharmaceutics 12, no. 10: 975. https://doi.org/10.3390/pharmaceutics12100975