当前位置:
X-MOL 学术
›
Ind. Eng. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Microscopic Simulation of Methane Adsorption in Organic Matter
Industrial & Engineering Chemistry Research ( IF 4.2 ) Pub Date : 2019-02-13 , DOI: 10.1021/acs.iecr.8b05762 Jianfei Zhao 1 , Zhouhua Wang 1 , Ping Guo 1
Industrial & Engineering Chemistry Research ( IF 4.2 ) Pub Date : 2019-02-13 , DOI: 10.1021/acs.iecr.8b05762 Jianfei Zhao 1 , Zhouhua Wang 1 , Ping Guo 1
Affiliation
|
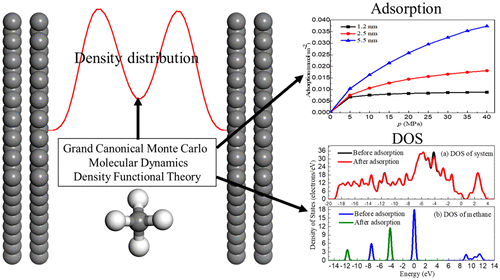
Shale is rich in nanopores with organic matter, but few studies have researched them in combination with molecular simulation and quantum mechanics. The organic matter was simplified into graphite and the graphene structure, and Grand Canonical Monte Carlo and Molecular Dynamics methods have been used to study methane adsorption in the different apertures of graphite; then, adsorption characteristics were calculated and analyzed by Density Functional Theory. The simulation results show that methane molecules form a tight adsorption layer near the wall with different orientations. The density distribution shows an obvious adsorption layer, and the interaction distance is 1 to 6 Å. The adsorption energy is distributed in the range of −0.2200 to −0.1080 eV, which states physical adsorption, and can be divided into two segments: 1–3 and 4–6 Å. Density of states (DOS) of systems are distributed within the range of −20 to 5 eV. II B5 and III B1 are different only at −5.0 to −3.6 eV, and DOS of adsorbents coincided before and after adsorption, while DOS of methane shift to the lower energy region after adsorption.
中文翻译:

甲烷在有机物中吸附的微观模拟
页岩富含有机物的纳米孔,但是很少有研究结合分子模拟和量子力学对其进行研究。将有机物简化为石墨和石墨烯结构,并使用Grand Canonical Monte Carlo和分子动力学方法研究了甲烷在不同孔径的石墨中的吸附。然后,通过密度泛函理论计算并分析了吸附特性。模拟结果表明,甲烷分子在壁附近以不同的方向形成了紧密的吸附层。密度分布显示出明显的吸附层,相互作用距离为1至6Å。吸附能分布在-0.2200至-0.1080 eV的范围内,它表示物理吸附,可以分为两个部分:1-3和4-6Å。系统的状态密度(DOS)分布在-20至5 eV的范围内。II B5和III B1仅在-5.0到-3.6 eV时不同,并且吸附剂的DOS吸附前后均重合,而甲烷的DOS在吸附后移至较低的能量区。
更新日期:2019-02-14
中文翻译:
甲烷在有机物中吸附的微观模拟
页岩富含有机物的纳米孔,但是很少有研究结合分子模拟和量子力学对其进行研究。将有机物简化为石墨和石墨烯结构,并使用Grand Canonical Monte Carlo和分子动力学方法研究了甲烷在不同孔径的石墨中的吸附。然后,通过密度泛函理论计算并分析了吸附特性。模拟结果表明,甲烷分子在壁附近以不同的方向形成了紧密的吸附层。密度分布显示出明显的吸附层,相互作用距离为1至6Å。吸附能分布在-0.2200至-0.1080 eV的范围内,它表示物理吸附,可以分为两个部分:1-3和4-6Å。系统的状态密度(DOS)分布在-20至5 eV的范围内。II B5和III B1仅在-5.0到-3.6 eV时不同,并且吸附剂的DOS吸附前后均重合,而甲烷的DOS在吸附后移至较低的能量区。

























 京公网安备 11010802027423号
京公网安备 11010802027423号