Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Performance of Density-Functional Tight-Binding in Comparison to Ab Initio and First-Principles Methods for Isomer Geometries and Energies of Glucose Epimers in Vacuo and Solution
ACS Omega ( IF 4.1 ) Pub Date : 2018-12-07 00:00:00 , DOI: 10.1021/acsomega.8b02213 Ka Hung Lee 1, 2 , Udo Schnupf 3 , Bobby G. Sumpter , Stephan Irle 1
ACS Omega ( IF 4.1 ) Pub Date : 2018-12-07 00:00:00 , DOI: 10.1021/acsomega.8b02213 Ka Hung Lee 1, 2 , Udo Schnupf 3 , Bobby G. Sumpter , Stephan Irle 1
Affiliation
|
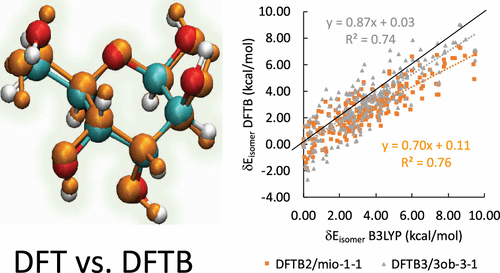
Density functional theory (DFT) is a widely used methodology for the computation of molecular and electronic structure, and we confirm that B3LYP and the high-level ab initio G3B3 method are in excellent agreement for the lowest-energy isomers of the 16 glucose epimers. Density-functional tight-binding (DFTB) is an approximate version of DFT with typically comparable accuracy that is 2 to 3 orders of magnitude faster, therefore generally very suitable for processing large numbers of complex structures. Conformational isomerism in sugars is well known to give rise to a large number of isomer structures. On the basis of a comprehensive study of glucose epimers in vacuo and aqueous solution, we found that the performance of DFTB is on par to B3LYP in terms of geometrical parameters excluding hydrogen bonds and isomer energies. However, DFTB underestimates both hydrogen bonding interactions as well as torsional barriers associated with rotations of the hydroxy groups, resulting in a counterintuitive overemphasis of hydrogen bonding in both gas phase as well as in water. Although the associated root mean squared deviation from B3LYP within epimer isomer groups is only on the order of 1 kcal/mol, this deviation affects the correct assignment of major isomer ordering, which span less than 10 kcal/mol. Both second- as well as third-order DFTB methods are exhibiting similar deviations from B3LYP. Even after the inclusion of empirical dispersion corrections in vacuum, these deviations remain for a large majority of isomer energies and geometries when compared to dispersion-corrected B3LYP.
中文翻译:

与Vauo和溶液中葡萄糖差向异构体的异构体几何构型和能量的从头算和第一性原理相比,密度功能紧密结合的性能
密度泛函理论(DFT)是一种广泛用于计算分子和电子结构的方法,我们确认B3LYP和高水平从头算G3B3方法与16种葡萄糖差向异构体的最低能量异构体完全吻合。密度功能紧密绑定(DFTB)是DFT的近似版本,其精度通常可比,快了2到3个数量级,因此通常非常适合处理大量复杂结构。众所周知,糖中的构象异构会产生大量的异构体结构。在对葡萄糖差向异构体在真空和水溶液中的综合研究的基础上,我们发现DFTB的性能在几何参数方面与B3LYP相当,但不包括氢键和异构体能。然而,DFTB低估了氢键相互作用以及与羟基旋转相关的扭转壁垒,导致气相和水中氢键的过分直觉都与直觉相反。尽管差向异构体中B3LYP的相关均方根偏差仅为1 kcal / mol,但这种偏差会影响主要异构体有序分布的正确分配,其分布范围小于10 kcal / mol。二阶和三阶DFTB方法都表现出与B3LYP相似的偏差。即使在真空中包括经验色散校正后,与色散校正的B3LYP相比,对于大多数异构体能量和几何形状,这些偏差仍然存在。
更新日期:2018-12-07
中文翻译:
与Vauo和溶液中葡萄糖差向异构体的异构体几何构型和能量的从头算和第一性原理相比,密度功能紧密结合的性能
密度泛函理论(DFT)是一种广泛用于计算分子和电子结构的方法,我们确认B3LYP和高水平从头算G3B3方法与16种葡萄糖差向异构体的最低能量异构体完全吻合。密度功能紧密绑定(DFTB)是DFT的近似版本,其精度通常可比,快了2到3个数量级,因此通常非常适合处理大量复杂结构。众所周知,糖中的构象异构会产生大量的异构体结构。在对葡萄糖差向异构体在真空和水溶液中的综合研究的基础上,我们发现DFTB的性能在几何参数方面与B3LYP相当,但不包括氢键和异构体能。然而,DFTB低估了氢键相互作用以及与羟基旋转相关的扭转壁垒,导致气相和水中氢键的过分直觉都与直觉相反。尽管差向异构体中B3LYP的相关均方根偏差仅为1 kcal / mol,但这种偏差会影响主要异构体有序分布的正确分配,其分布范围小于10 kcal / mol。二阶和三阶DFTB方法都表现出与B3LYP相似的偏差。即使在真空中包括经验色散校正后,与色散校正的B3LYP相比,对于大多数异构体能量和几何形状,这些偏差仍然存在。
























 京公网安备 11010802027423号
京公网安备 11010802027423号