当前位置:
X-MOL 学术
›
J. Biomed. Inform.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A topological approach for cancer subtyping from gene expression data.
Journal of Biomedical informatics ( IF 4.5 ) Pub Date : 2019-12-29 , DOI: 10.1016/j.jbi.2019.103357 Omar Rafique 1 , A H Mir 2
Journal of Biomedical informatics ( IF 4.5 ) Pub Date : 2019-12-29 , DOI: 10.1016/j.jbi.2019.103357 Omar Rafique 1 , A H Mir 2
Affiliation
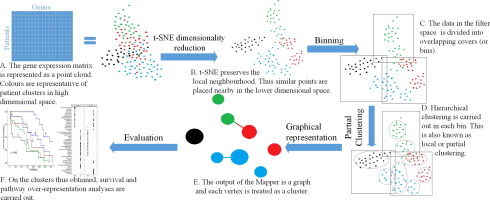
|
BACKGROUND
Gene expression data contains key information which can be used for subtyping cancer patients. However, computational methods suffer from 'curse of dimensionality' due to very high dimensionality of omics data and therefore are not able to clearly distinguish between the discovered subtypes in terms of separation of survival plots.
METHODS
To address this we propose a framework based on Topological Mapper algorithm. The novelty of this work is that we suggest a method for defining the filter function on which the mapper algorithm heavily depends. Survival analysis of the discovered cancer subtypes is carried out and evaluated in terms of minimum pairwise separation between the Kaplan-Meier plots. Furthermore, we present a method to measure the separation between the discovered subtypes based on hazard ratios.
RESULTS
Five cancer genomics datasets obtained from The Cancer Genome Atlas portal have been used for comparisons with Robust Sparse Correlation-Otrimle (RSC-Otrimle) algorithm and Similarity Network Fusion(SNF). Comparisons show that the minimum pairwise life expectancy difference (in days) between the discovered subtypes for lung, colon, breast, glioblastoma and kidney cancers is 107, 204, 20, 88 and 425 days, respectively, for the proposed methodology whereas it is only 69, 43, 6, 61 and 282 days for RSC-Otrimle and 9, 95, 18, 60 and 148 days for SNF. Hazard ratio analysis also shows that the proposed methodology performs better in four of the five datasets. A visual inspection of Kaplan-Meier plots reveals that the proposed methodology achieves lesser overlap in Kaplan-Meier plots especially for lung, breast and kidney cases. Furthermore, relevant genetic pathways for each subtype have been obtained and pathways which can be possible targets for treatment have been discussed.
CONCLUSION
The significance of this work lies in individualized understanding of cancer from patient to patient which is the backbone of Precision Medicine.
中文翻译:

从基因表达数据进行癌症分型的一种拓扑方法。
背景技术基因表达数据包含可用于分型癌症患者的关键信息。但是,由于组学数据的维数很高,因此计算方法会遭受“维数诅咒”,因此无法根据生存图的分离来清楚地区分所发现的子类型。方法为了解决这个问题,我们提出了一个基于拓扑映射器算法的框架。这项工作的新颖性在于,我们提出了一种定义映射器算法严重依赖的滤波函数的方法。进行发现的癌症亚型的生存分析,并根据Kaplan-Meier图之间的最小成对间隔进行评估。此外,我们提出了一种基于风险比来衡量发现的亚型之间的分离的方法。结果从癌症基因组图集门户网站获得的五个癌症基因组数据已用于与鲁棒稀疏相关算法(RSC-Otrimle)算法和相似网络融合(SNF)进行比较。比较表明,对于拟议的方法,发现的肺癌,结肠癌,乳腺癌,成胶质细胞瘤和肾癌亚型之间的最小成对预期寿命差异(以天为单位)分别为107、204、20、88和425天,而这仅是RSC-Otrimle为69、43、6、61和282天,SNF为9、95、18、60和148天。危险比分析还表明,所提出的方法在五个数据集中的四个中表现更好。目视检查Kaplan-Meier图时发现,所提出的方法在Kaplan-Meier图中实现了较少的重叠,特别是对于肺,乳腺和肾脏病例。此外,已经获得了每种亚型的相关遗传途径,并讨论了可能成为治疗靶点的途径。结论这项工作的意义在于从个体到个体对癌症的个性化理解,这是精准医学的基础。
更新日期:2019-12-29
中文翻译:
从基因表达数据进行癌症分型的一种拓扑方法。
背景技术基因表达数据包含可用于分型癌症患者的关键信息。但是,由于组学数据的维数很高,因此计算方法会遭受“维数诅咒”,因此无法根据生存图的分离来清楚地区分所发现的子类型。方法为了解决这个问题,我们提出了一个基于拓扑映射器算法的框架。这项工作的新颖性在于,我们提出了一种定义映射器算法严重依赖的滤波函数的方法。进行发现的癌症亚型的生存分析,并根据Kaplan-Meier图之间的最小成对间隔进行评估。此外,我们提出了一种基于风险比来衡量发现的亚型之间的分离的方法。结果从癌症基因组图集门户网站获得的五个癌症基因组数据已用于与鲁棒稀疏相关算法(RSC-Otrimle)算法和相似网络融合(SNF)进行比较。比较表明,对于拟议的方法,发现的肺癌,结肠癌,乳腺癌,成胶质细胞瘤和肾癌亚型之间的最小成对预期寿命差异(以天为单位)分别为107、204、20、88和425天,而这仅是RSC-Otrimle为69、43、6、61和282天,SNF为9、95、18、60和148天。危险比分析还表明,所提出的方法在五个数据集中的四个中表现更好。目视检查Kaplan-Meier图时发现,所提出的方法在Kaplan-Meier图中实现了较少的重叠,特别是对于肺,乳腺和肾脏病例。此外,已经获得了每种亚型的相关遗传途径,并讨论了可能成为治疗靶点的途径。结论这项工作的意义在于从个体到个体对癌症的个性化理解,这是精准医学的基础。


























 京公网安备 11010802027423号
京公网安备 11010802027423号