当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Implementation of Geometry-Dependent Charge Flux into the Polarizable AMOEBA+ Potential.
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2019-12-22 , DOI: 10.1021/acs.jpclett.9b03489 Chengwen Liu 1 , Jean-Philip Piquemal 1, 2, 3 , Pengyu Ren 1
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2019-12-22 , DOI: 10.1021/acs.jpclett.9b03489 Chengwen Liu 1 , Jean-Philip Piquemal 1, 2, 3 , Pengyu Ren 1
Affiliation
|
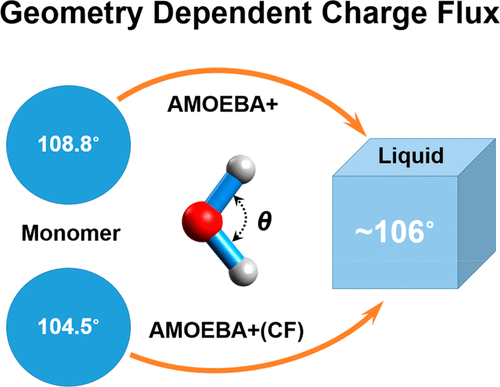
Molecular dynamics (MD) simulations employing classical force fields (FFs) have been widely used to model molecular systems. The important ingredient of the current FFs, atomic charge, remains fixed during MD simulations despite the atomic environment or local geometry changes. This approximation hinders the transferability of the potential being used in multiple phases. Here we implement a geometry-dependent charge flux (GDCF) model into the multipole-based AMOEBA+ polarizable potential. The CF in the current work explicitly depends on the local geometry (bond and angle) of the molecule. To our knowledge, this is the first study that derives energy and force expressions due to GDCF in a multipole-based polarizable FF framework. Due to the inclusion of GDCF, the AMOEBA+ water model is noticeably improved in terms of describing the monomer properties, cluster binding/interaction energy, and a variety of liquid properties, including the infrared spectra that previous flexible water models were not able to capture.
中文翻译:

将几何相关的电荷通量实现到极化AMOEBA +电位中。
使用经典力场(FFs)的分子动力学(MD)模拟已被广泛用于对分子系统进行建模。尽管原子环境或局部几何形状发生变化,但当前FF的重要成分原子电荷在MD模拟过程中保持固定。这种近似阻碍了在多相中使用的电势的可转移性。在这里,我们将基于几何的电荷通量(GDCF)模型实现到基于多极的AMOEBA +可极化电势中。当前工作中的CF明显取决于分子的局部几何形状(键和角度)。据我们所知,这是第一个在基于多极的可极化FF框架中导出GDCF引起的能量和力表达的研究。由于包含了GDCF,
更新日期:2019-12-31
中文翻译:
将几何相关的电荷通量实现到极化AMOEBA +电位中。
使用经典力场(FFs)的分子动力学(MD)模拟已被广泛用于对分子系统进行建模。尽管原子环境或局部几何形状发生变化,但当前FF的重要成分原子电荷在MD模拟过程中保持固定。这种近似阻碍了在多相中使用的电势的可转移性。在这里,我们将基于几何的电荷通量(GDCF)模型实现到基于多极的AMOEBA +可极化电势中。当前工作中的CF明显取决于分子的局部几何形状(键和角度)。据我们所知,这是第一个在基于多极的可极化FF框架中导出GDCF引起的能量和力表达的研究。由于包含了GDCF,


























 京公网安备 11010802027423号
京公网安备 11010802027423号