当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
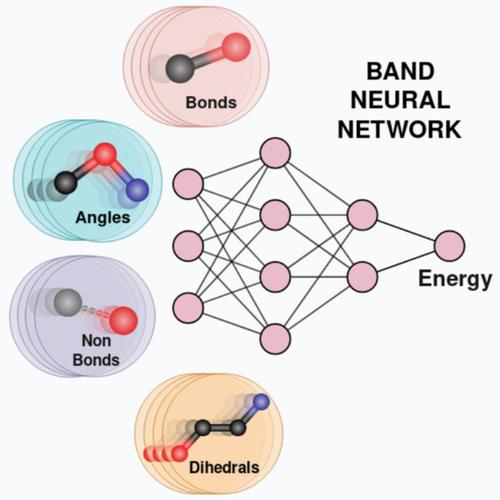
BAND NN: A Deep Learning Framework for Energy Prediction and Geometry Optimization of Organic Small Molecules
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2019-12-17 , DOI: 10.1002/jcc.26128 Siddhartha Laghuvarapu 1 , Yashaswi Pathak 1 , U Deva Priyakumar 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2019-12-17 , DOI: 10.1002/jcc.26128 Siddhartha Laghuvarapu 1 , Yashaswi Pathak 1 , U Deva Priyakumar 1
Affiliation
|
Recent advances in artificial intelligence along with the development of large data sets of energies calculated using quantum mechanical (QM)/density functional theory (DFT) methods have enabled prediction of accurate molecular energies at reasonably low computational cost. However, machine learning models that have been reported so far require the atomic positions obtained from geometry optimizations using high‐level QM/DFT methods as input in order to predict the energies and do not allow for geometry optimization. In this study, a transferable and molecule size‐independent machine learning model bonds (B), angles (A), nonbonded (N) interactions, and dihedrals (D) neural network (BAND NN) based on a chemically intuitive representation inspired by molecular mechanics force fields is presented. The model predicts the atomization energies of equilibrium and nonequilibrium structures as sum of energy contributions from bonds (B), angles (A), nonbonds (N), and dihedrals (D) at remarkable accuracy. The robustness of the proposed model is further validated by calculations that span over the conformational, configurational, and reaction space. The transferability of this model on systems larger than the ones in the data set is demonstrated by performing calculations on selected large molecules. Importantly, employing the BAND NN model, it is possible to perform geometry optimizations starting from nonequilibrium structures along with predicting their energies. © 2019 Wiley Periodicals, Inc.
中文翻译:

BAND NN:有机小分子能量预测和几何优化的深度学习框架
人工智能的最新进展以及使用量子力学 (QM)/密度泛函理论 (DFT) 方法计算的大型能量数据集的发展,使得能够以相当低的计算成本预测准确的分子能量。然而,迄今为止报道的机器学习模型需要使用高级 QM/DFT 方法从几何优化中获得的原子位置作为输入,以预测能量,并且不允许几何优化。在这项研究中,一个可转移且与分子大小无关的机器学习模型基于受分子启发的化学直观表示,结合 (B)、角度 (A)、非键 (N) 相互作用和二面角 (D) 神经网络 (BAND NN)。力学力场。该模型将平衡和非平衡结构的原子化能预测为来自键 (B)、角度 (A)、非键 (N) 和二面角 (D) 的能量贡献的总和。通过跨越构象、构型和反应空间的计算进一步验证了所提出模型的稳健性。通过对选定的大分子进行计算,证明了该模型在比数据集中的系统更大的系统上的可转移性。重要的是,使用 BAND NN 模型,可以从非平衡结构开始执行几何优化,同时预测它们的能量。© 2019 威利期刊公司。通过跨越构象、构型和反应空间的计算进一步验证了所提出模型的稳健性。通过对选定的大分子进行计算,证明了该模型在比数据集中的系统更大的系统上的可转移性。重要的是,使用 BAND NN 模型,可以从非平衡结构开始执行几何优化,同时预测它们的能量。© 2019 威利期刊公司。通过跨越构象、构型和反应空间的计算进一步验证了所提出模型的稳健性。通过对选定的大分子进行计算,证明了该模型在大于数据集中的系统上的可转移性。重要的是,使用 BAND NN 模型,可以从非平衡结构开始执行几何优化,同时预测它们的能量。© 2019 威利期刊公司。可以从非平衡结构开始进行几何优化,同时预测它们的能量。© 2019 威利期刊公司。可以从非平衡结构开始进行几何优化,同时预测它们的能量。© 2019 威利期刊公司。
更新日期:2019-12-17
中文翻译:
BAND NN:有机小分子能量预测和几何优化的深度学习框架
人工智能的最新进展以及使用量子力学 (QM)/密度泛函理论 (DFT) 方法计算的大型能量数据集的发展,使得能够以相当低的计算成本预测准确的分子能量。然而,迄今为止报道的机器学习模型需要使用高级 QM/DFT 方法从几何优化中获得的原子位置作为输入,以预测能量,并且不允许几何优化。在这项研究中,一个可转移且与分子大小无关的机器学习模型基于受分子启发的化学直观表示,结合 (B)、角度 (A)、非键 (N) 相互作用和二面角 (D) 神经网络 (BAND NN)。力学力场。该模型将平衡和非平衡结构的原子化能预测为来自键 (B)、角度 (A)、非键 (N) 和二面角 (D) 的能量贡献的总和。通过跨越构象、构型和反应空间的计算进一步验证了所提出模型的稳健性。通过对选定的大分子进行计算,证明了该模型在比数据集中的系统更大的系统上的可转移性。重要的是,使用 BAND NN 模型,可以从非平衡结构开始执行几何优化,同时预测它们的能量。© 2019 威利期刊公司。通过跨越构象、构型和反应空间的计算进一步验证了所提出模型的稳健性。通过对选定的大分子进行计算,证明了该模型在比数据集中的系统更大的系统上的可转移性。重要的是,使用 BAND NN 模型,可以从非平衡结构开始执行几何优化,同时预测它们的能量。© 2019 威利期刊公司。通过跨越构象、构型和反应空间的计算进一步验证了所提出模型的稳健性。通过对选定的大分子进行计算,证明了该模型在大于数据集中的系统上的可转移性。重要的是,使用 BAND NN 模型,可以从非平衡结构开始执行几何优化,同时预测它们的能量。© 2019 威利期刊公司。可以从非平衡结构开始进行几何优化,同时预测它们的能量。© 2019 威利期刊公司。可以从非平衡结构开始进行几何优化,同时预测它们的能量。© 2019 威利期刊公司。

























 京公网安备 11010802027423号
京公网安备 11010802027423号