当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
SPOT‐Fold: Fragment‐Free Protein Structure Prediction Guided by Predicted Backbone Structure and Contact Map
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2019-12-17 , DOI: 10.1002/jcc.26132 Yufeng Cai 1 , Xiongjun Li 1 , Zhe Sun 1 , Yutong Lu 1 , Huiying Zhao 2 , Jack Hanson 3 , Kuldip Paliwal 3 , Thomas Litfin 4 , Yaoqi Zhou 4 , Yuedong Yang 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2019-12-17 , DOI: 10.1002/jcc.26132 Yufeng Cai 1 , Xiongjun Li 1 , Zhe Sun 1 , Yutong Lu 1 , Huiying Zhao 2 , Jack Hanson 3 , Kuldip Paliwal 3 , Thomas Litfin 4 , Yaoqi Zhou 4 , Yuedong Yang 1
Affiliation
|
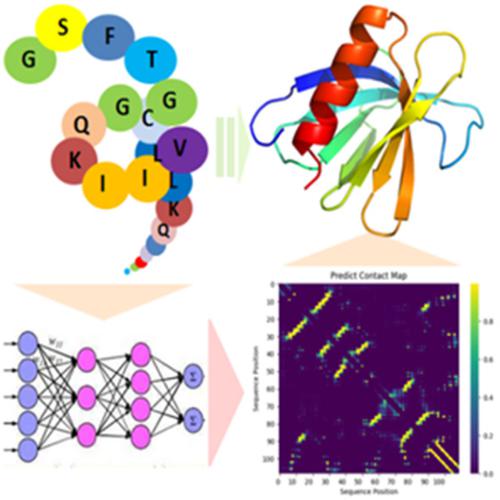
Protein structure determination has long been one of the most challenging problems in molecular biology for the past 60 years. Here we present an ab initio protein tertiary‐structure prediction method assisted by predicted contact maps from SPOT‐Contact and predicted dihedral angles from SPIDER 3. These predicted properties were then fed to the crystallography and NMR system (CNS) for restrained structure modeling. The resulted structures are first evaluated by the potential energy calculated by CNS, followed by dDFIRE energy function for model selections. The method called SPOT‐Fold has been tested on 241 CASP targets between 67 and 670 amino acid residues, 60 randomly selected globular proteins under 100 amino acids. The method has a comparable accuracy to other contact‐map‐based modeling techniques. © 2019 Wiley Periodicals, Inc.
中文翻译:

SPOT-Fold:由预测骨架结构和接触图引导的无片段蛋白质结构预测
蛋白质结构测定长期以来一直是过去 60 年来分子生物学中最具挑战性的问题之一。在这里,我们提出了一种由 SPOT-Contact 预测的接触图和 SPIDER 3 预测的二面角辅助的从头开始的蛋白质三级结构预测方法。然后将这些预测的特性输入晶体学和 NMR 系统 (CNS) 以进行约束结构建模。所得结构首先通过 CNS 计算的势能进行评估,然后是用于模型选择的 dDFIRE 能量函数。这种名为 SPOT-Fold 的方法已经在 67 到 670 个氨基酸残基之间的 241 个 CASP 靶标、100 个氨基酸以下的 60 个随机选择的球状蛋白质上进行了测试。该方法具有与其他基于接触图的建模技术相当的准确性。© 2019 威利期刊公司。
更新日期:2019-12-17
中文翻译:
SPOT-Fold:由预测骨架结构和接触图引导的无片段蛋白质结构预测
蛋白质结构测定长期以来一直是过去 60 年来分子生物学中最具挑战性的问题之一。在这里,我们提出了一种由 SPOT-Contact 预测的接触图和 SPIDER 3 预测的二面角辅助的从头开始的蛋白质三级结构预测方法。然后将这些预测的特性输入晶体学和 NMR 系统 (CNS) 以进行约束结构建模。所得结构首先通过 CNS 计算的势能进行评估,然后是用于模型选择的 dDFIRE 能量函数。这种名为 SPOT-Fold 的方法已经在 67 到 670 个氨基酸残基之间的 241 个 CASP 靶标、100 个氨基酸以下的 60 个随机选择的球状蛋白质上进行了测试。该方法具有与其他基于接触图的建模技术相当的准确性。© 2019 威利期刊公司。

























 京公网安备 11010802027423号
京公网安备 11010802027423号