当前位置:
X-MOL 学术
›
J. Chin. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Design of antimalarial agents based on pyrimidine derivatives as methionine aminopeptidase 1b inhibitor: Molecular docking, quantitative structure activity relationships, and molecular dynamics simulation studies
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2019-12-17 , DOI: 10.1002/jccs.201900165 Maryam Iman 1 , Asghar Davood 2 , Ali Khamesipour 3
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2019-12-17 , DOI: 10.1002/jccs.201900165 Maryam Iman 1 , Asghar Davood 2 , Ali Khamesipour 3
Affiliation
|
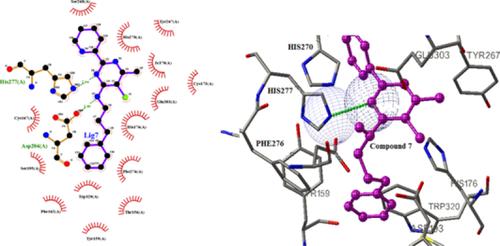
Malaria is a parasitic disease mainly caused by Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, and Plasmodium ovale and rarely by animal plasmodium such as Plasmodium knowlesi. Despite the worldwide endorsement of eradicating malaria, the mortality and morbidity of malaria is still high, especially in children of resource‐poor regions of the world. In the current study, a series of 2‐pyridyl pyrimidine derivatives with P. falciparum methionine aminopeptidase 1b (MA1b) inhibitory activity were subjected to quantitative structure, relationship activities, molecular docking, and molecular dynamics simulation to identify the ideal physicochemical characteristics and the drug–receptor interactions of potential antimalarial compounds. The structures were built using HyperChem. A docking study was performed using AutoDock. The obtained quantitative structure activity relationships model has showed that the descriptors MLOGP, E2p, Mor24u, PW2, and H8m mainly affect the antimalarial activity of the series of ligands. The residues His 277, Fe 370, Asp 240, Trp 320, and Tyr 260 of MA1b play a very important role in the drug–enzyme interaction by creating hydrogen bond, π‐cation, and π‐π interactions. To give a complete impression about the pyrimidine derivatives, compound 7 was subjected to 1,000 ps molecular dynamics (MD) simulations. The best structure based on our study was suggested for a future new drug with potential antimalarial compounds.
中文翻译:

基于嘧啶衍生物作为蛋氨酸氨基肽酶1b抑制剂的抗疟药的设计:分子对接,定量结构活性关系和分子动力学模拟研究
疟疾是一种寄生性疾病主要是由恶性疟原虫,间日疟原虫,三日疟原虫,和卵形疟原虫,很少由动物疟原虫诸如诺氏疟原虫。尽管全世界都认可消除疟疾,但疟疾的死亡率和发病率仍然很高,尤其是在世界资源贫乏地区的儿童中。在当前的研究中,一系列的2-吡啶基嘧啶衍生物与恶性疟原虫对甲硫氨酸氨基肽酶1b(MA1b)的抑制活性进行了定量结构,关联活性,分子对接和分子动力学模拟,以鉴定理想的理化特性和潜在抗疟疾化合物的药物-受体相互作用。这些结构是使用HyperChem构建的。使用AutoDock进行了对接研究。获得的定量结构活性关系模型表明,描述符MLOGP,E2p,Mor24u,PW2和H8m主要影响该系列配体的抗疟活性。MA1b的His 277,Fe 370,Asp 240,Trp 320和Tyr 260残基通过建立氢键,π-阳离子和π-π相互作用,在药物-酶相互作用中起着非常重要的作用。为了给人一个关于嘧啶衍生物的完整印象,对化合物7进行了1,000 ps的分子动力学(MD)模拟。根据我们的研究,最好的结构被建议用于未来的具有潜在抗疟疾化合物的新药物。
更新日期:2019-12-17
中文翻译:
基于嘧啶衍生物作为蛋氨酸氨基肽酶1b抑制剂的抗疟药的设计:分子对接,定量结构活性关系和分子动力学模拟研究
疟疾是一种寄生性疾病主要是由恶性疟原虫,间日疟原虫,三日疟原虫,和卵形疟原虫,很少由动物疟原虫诸如诺氏疟原虫。尽管全世界都认可消除疟疾,但疟疾的死亡率和发病率仍然很高,尤其是在世界资源贫乏地区的儿童中。在当前的研究中,一系列的2-吡啶基嘧啶衍生物与恶性疟原虫对甲硫氨酸氨基肽酶1b(MA1b)的抑制活性进行了定量结构,关联活性,分子对接和分子动力学模拟,以鉴定理想的理化特性和潜在抗疟疾化合物的药物-受体相互作用。这些结构是使用HyperChem构建的。使用AutoDock进行了对接研究。获得的定量结构活性关系模型表明,描述符MLOGP,E2p,Mor24u,PW2和H8m主要影响该系列配体的抗疟活性。MA1b的His 277,Fe 370,Asp 240,Trp 320和Tyr 260残基通过建立氢键,π-阳离子和π-π相互作用,在药物-酶相互作用中起着非常重要的作用。为了给人一个关于嘧啶衍生物的完整印象,对化合物7进行了1,000 ps的分子动力学(MD)模拟。根据我们的研究,最好的结构被建议用于未来的具有潜在抗疟疾化合物的新药物。

























 京公网安备 11010802027423号
京公网安备 11010802027423号