当前位置:
X-MOL 学术
›
Inorg. Chem. Front.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Insights into the complexity of the excited states of Eu-doped luminescent materials
Inorganic Chemistry Frontiers ( IF 7 ) Pub Date : 2019/12/17 , DOI: 10.1039/c9qi01455a Jonas J. Joos 1, 2, 3, 4, 5 , Philippe F. Smet 1, 2, 3, 4, 5 , Luis Seijo 6, 7, 8, 9, 10 , Zoila Barandiarán 6, 7, 8, 9, 10
Inorganic Chemistry Frontiers ( IF 7 ) Pub Date : 2019/12/17 , DOI: 10.1039/c9qi01455a Jonas J. Joos 1, 2, 3, 4, 5 , Philippe F. Smet 1, 2, 3, 4, 5 , Luis Seijo 6, 7, 8, 9, 10 , Zoila Barandiarán 6, 7, 8, 9, 10
Affiliation
|
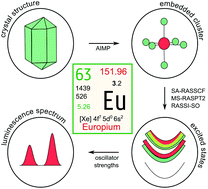
It has always been a spectroscopist's dream to correlate a material's luminescence properties with its microscopic structure, based on reliable structure–property relationships. Electronic structure methods are promising to achieve this goal; yet they are especially challenging in the case of Eu-based materials which are known to feature exceptionally high density of excited states, large spins and severe electron correlation. In this work, state-of-the-art multiconfigurational ab initio embedded-cluster methods are applied to gain a deeper insight into the luminescence mechanisms of Eu2+ and Eu3+-doped phosphors. Regardless of the difficulties, very accurate excitation energies are achieved, reaching 68% prediction intervals of 300 cm−1, corresponding to an accuracy of 5–10 nm in the visible wavelength range. Complete configurational coordinate curves are obtained, yielding breathing mode vibrational frequencies and equilibrium bond lengths for all excited states. Moreover, electric dipole transition moments and oscillator strengths are used to calculate absorption spectra. Excellent agreement with experiment is found. The ab initio calculations give an unprecedented detailed view of the Eu2+ excited state landscape, allowing for an improved understanding of its structure, including the origin of the so-called ‘staircase structure’ and the role of ligand covalency in the ligand field and exchange splitting. It is found that more covalent host compounds feature higher exchange splittings due to an increased stabilization of high-spin states by the interaction with virtual LMCT states. It is verified that the equilibrium Eu–ligand bond length contracts upon 4f–5d excitation towards the lowest 5d submanifold and that the bond lengths are directly related to the configurational character of the electronic eigenstate. Moreover, comparing ab initio calculations with crystal field calculations proves that a decoupled model for the Eu2+ excited states is inadequate, making the use of an intermediate coupling scheme compulsory. With this approach, computational design of luminescent materials is getting within reach.
中文翻译:

洞悉Eu掺杂发光材料激发态的复杂性
基于可靠的结构-属性关系,将材料的发光特性与其微观结构相关联,一直是光谱学家的梦想。电子结构方法有望实现这一目标。然而,对于以Eu基材料为例,这些材料尤为具有挑战性,因为Eu基材料具有异常高的激发态密度,大的自旋和严重的电子相关性。在这项工作中,采用了最先进的多配置从头开始嵌入簇方法,以更深入地了解Eu 2+和Eu 3+掺杂磷光体的发光机理。无论困难如何,都能获得非常精确的激发能,达到300 cm -1的68%预测间隔,对应于可见波长范围内5–10 nm的精度。获得了完整的构型坐标曲线,得出了所有激发态的呼吸模式振动频率和平衡键长。此外,电偶极跃迁矩和振荡器强度可用于计算吸收光谱。发现与实验极好的一致性。该从头计算给出一个欧盟前所未有的详细视图2+激发态景观,可以更好地了解其结构,包括所谓“楼梯结构”的起源以及配体共价在配体场和交换分裂中的作用。发现通过与虚拟LMCT状态的相互作用,由于高自旋态的增加的稳定性,更多的共价主体化合物具有较高的交换分裂。证实了平衡Eu-配体键长在4f-5d激发时向最低5d子流形收缩,并且键长与电子本征态的结构特征直接相关。此外,从头算与晶体场计算的比较证明了Eu 2+的解耦模型激发态不足,因此必须强制使用中间耦合方案。通过这种方法,发光材料的计算设计已经触手可及。
更新日期:2020-02-18
中文翻译:
洞悉Eu掺杂发光材料激发态的复杂性
基于可靠的结构-属性关系,将材料的发光特性与其微观结构相关联,一直是光谱学家的梦想。电子结构方法有望实现这一目标。然而,对于以Eu基材料为例,这些材料尤为具有挑战性,因为Eu基材料具有异常高的激发态密度,大的自旋和严重的电子相关性。在这项工作中,采用了最先进的多配置从头开始嵌入簇方法,以更深入地了解Eu 2+和Eu 3+掺杂磷光体的发光机理。无论困难如何,都能获得非常精确的激发能,达到300 cm -1的68%预测间隔,对应于可见波长范围内5–10 nm的精度。获得了完整的构型坐标曲线,得出了所有激发态的呼吸模式振动频率和平衡键长。此外,电偶极跃迁矩和振荡器强度可用于计算吸收光谱。发现与实验极好的一致性。该从头计算给出一个欧盟前所未有的详细视图2+激发态景观,可以更好地了解其结构,包括所谓“楼梯结构”的起源以及配体共价在配体场和交换分裂中的作用。发现通过与虚拟LMCT状态的相互作用,由于高自旋态的增加的稳定性,更多的共价主体化合物具有较高的交换分裂。证实了平衡Eu-配体键长在4f-5d激发时向最低5d子流形收缩,并且键长与电子本征态的结构特征直接相关。此外,从头算与晶体场计算的比较证明了Eu 2+的解耦模型激发态不足,因此必须强制使用中间耦合方案。通过这种方法,发光材料的计算设计已经触手可及。

























 京公网安备 11010802027423号
京公网安备 11010802027423号