当前位置:
X-MOL 学术
›
PLOS Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Leveraging allelic imbalance to refine fine-mapping for eQTL studies.
PLOS Genetics ( IF 4.5 ) Pub Date : 2019-12-13 , DOI: 10.1371/journal.pgen.1008481 Jennifer Zou 1 , Farhad Hormozdiari 2, 3 , Brandon Jew 4 , Stephane E Castel 5, 6 , Tuuli Lappalainen 5, 6 , Jason Ernst 1, 7 , Jae Hoon Sul 8 , Eleazar Eskin 1, 9
PLOS Genetics ( IF 4.5 ) Pub Date : 2019-12-13 , DOI: 10.1371/journal.pgen.1008481 Jennifer Zou 1 , Farhad Hormozdiari 2, 3 , Brandon Jew 4 , Stephane E Castel 5, 6 , Tuuli Lappalainen 5, 6 , Jason Ernst 1, 7 , Jae Hoon Sul 8 , Eleazar Eskin 1, 9
Affiliation
|
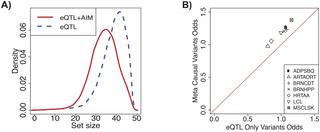
Many disease risk loci identified in genome-wide association studies are present in non-coding regions of the genome. Previous studies have found enrichment of expression quantitative trait loci (eQTLs) in disease risk loci, indicating that identifying causal variants for gene expression is important for elucidating the genetic basis of not only gene expression but also complex traits. However, detecting causal variants is challenging due to complex genetic correlation among variants known as linkage disequilibrium (LD) and the presence of multiple causal variants within a locus. Although several fine-mapping approaches have been developed to overcome these challenges, they may produce large sets of putative causal variants when true causal variants are in high LD with many non-causal variants. In eQTL studies, there is an additional source of information that can be used to improve fine-mapping called allelic imbalance (AIM) that measures imbalance in gene expression on two chromosomes of a diploid organism. In this work, we develop a novel statistical method that leverages both AIM and total expression data to detect causal variants that regulate gene expression. We illustrate through simulations and application to 10 tissues of the Genotype-Tissue Expression (GTEx) dataset that our method identifies the true causal variants with higher specificity than an approach that uses only eQTL information. Across all tissues and genes, our method achieves a median reduction rate of 11% in the number of putative causal variants. We use chromatin state data from the Roadmap Epigenomics Consortium to show that the putative causal variants identified by our method are enriched for active regions of the genome, providing orthogonal support that our method identifies causal variants with increased specificity.
中文翻译:

利用等位基因失衡来完善eQTL研究的精细映射。
在全基因组关联研究中确定的许多疾病风险基因座存在于基因组的非编码区域。先前的研究发现疾病风险基因座中表达定量性状基因座(eQTL)的丰富,表明鉴定基因表达的因果变体对于阐明基因表达和复杂性状的遗传基础都很重要。但是,由于因连锁不平衡(LD)而导致的变体之间复杂的遗传相关性以及一个基因座中存在多个因果变体,因此检测因果变体具有挑战性。尽管已经开发了几种精细映射方法来克服这些挑战,但是当真实因果变量处于高LD且有许多非因果变量时,它们可能会产生大量推定的因果变量。在eQTL研究中,还有一个可用于改善精细映射的信息来源,即等位基因失衡(AIM),用于测量二倍体生物体两个染色体上基因表达的失衡。在这项工作中,我们开发了一种新颖的统计方法,该方法利用了AIM和总表达数据来检测调节基因表达的因果变体。通过对10个组织的基因型组织表达(GTEx)数据集进行仿真和应用,我们的方法比仅使用eQTL信息的方法具有更高的特异性,从而可以识别出真正的因果变体。在所有组织和基因中,我们的方法在假定的因果变异数上实现了11%的中值降低率。
更新日期:2019-12-17
中文翻译:
利用等位基因失衡来完善eQTL研究的精细映射。
在全基因组关联研究中确定的许多疾病风险基因座存在于基因组的非编码区域。先前的研究发现疾病风险基因座中表达定量性状基因座(eQTL)的丰富,表明鉴定基因表达的因果变体对于阐明基因表达和复杂性状的遗传基础都很重要。但是,由于因连锁不平衡(LD)而导致的变体之间复杂的遗传相关性以及一个基因座中存在多个因果变体,因此检测因果变体具有挑战性。尽管已经开发了几种精细映射方法来克服这些挑战,但是当真实因果变量处于高LD且有许多非因果变量时,它们可能会产生大量推定的因果变量。在eQTL研究中,还有一个可用于改善精细映射的信息来源,即等位基因失衡(AIM),用于测量二倍体生物体两个染色体上基因表达的失衡。在这项工作中,我们开发了一种新颖的统计方法,该方法利用了AIM和总表达数据来检测调节基因表达的因果变体。通过对10个组织的基因型组织表达(GTEx)数据集进行仿真和应用,我们的方法比仅使用eQTL信息的方法具有更高的特异性,从而可以识别出真正的因果变体。在所有组织和基因中,我们的方法在假定的因果变异数上实现了11%的中值降低率。


























 京公网安备 11010802027423号
京公网安备 11010802027423号