当前位置:
X-MOL 学术
›
PLOS Negl. Trop. Dis.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Genomes of Leishmania parasites directly sequenced from patients with visceral leishmaniasis in the Indian subcontinent.
PLOS Neglected Tropical Diseases ( IF 3.8 ) Pub Date : 2019-12-12 , DOI: 10.1371/journal.pntd.0007900 Malgorzata A Domagalska 1 , Hideo Imamura 1 , Mandy Sanders 2 , Frederik Van den Broeck 1 , Narayan Raj Bhattarai 3 , Manu Vanaerschot 1 , Ilse Maes 1 , Erika D'Haenens 1 , Keshav Rai 3 , Suman Rijal 3 , Matthew Berriman 2 , James A Cotton 2 , Jean-Claude Dujardin 1, 4
PLOS Neglected Tropical Diseases ( IF 3.8 ) Pub Date : 2019-12-12 , DOI: 10.1371/journal.pntd.0007900 Malgorzata A Domagalska 1 , Hideo Imamura 1 , Mandy Sanders 2 , Frederik Van den Broeck 1 , Narayan Raj Bhattarai 3 , Manu Vanaerschot 1 , Ilse Maes 1 , Erika D'Haenens 1 , Keshav Rai 3 , Suman Rijal 3 , Matthew Berriman 2 , James A Cotton 2 , Jean-Claude Dujardin 1, 4
Affiliation
|
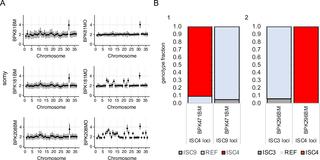
Whole genome sequencing (WGS) is increasingly used for molecular diagnosis and epidemiology of infectious diseases. Current Leishmania genomic studies rely on DNA extracted from cultured parasites, which might introduce sampling and biological biases into the subsequent analyses. Up to now, direct analysis of Leishmania genome in clinical samples is hampered by high levels of human DNA and large variation in parasite load in clinical samples. Here, we present a method, based on target enrichment of Leishmania donovani DNA with Agilent SureSelect technology, that allows the analysis of Leishmania genomes directly in clinical samples. We validated our protocol with a set of artificially mixed samples, followed by the analysis of 63 clinical samples (bone marrow or spleen aspirates) from visceral leishmaniasis patients in Nepal. We were able to identify genotypes using a set of diagnostic SNPs in almost all of these samples (97%) and access comprehensive genome-wide information in most (83%). This allowed us to perform phylogenomic analysis, assess chromosome copy number and identify large copy number variants (CNVs). Pairwise comparisons between the parasite genomes in clinical samples and derived in vitro cultured promastigotes showed a lower aneuploidy in amastigotes as well as genomic differences, suggesting polyclonal infections in patients. Altogether our results underline the need for sequencing parasite genomes directly in the host samples.
中文翻译:

直接从印度次大陆患有内脏利什曼病的患者中测序利什曼原虫寄生虫的基因组。
全基因组测序(WGS)越来越多地用于传染病的分子诊断和流行病学。当前的利什曼原虫基因组研究依赖于从培养的寄生虫中提取的DNA,这可能会在随后的分析中引入采样和生物学偏见。到目前为止,临床样品中利什曼原虫基因组的直接分析受到人类DNA含量高和临床样品中寄生虫载量变化大的困扰。在这里,我们介绍一种基于具有安捷伦SureSelect技术的多形利什曼原虫DNA靶标富集的方法,该方法可直接在临床样品中分析利什曼原虫基因组。我们使用一组人工混合的样本验证了我们的方案,然后分析了尼泊尔内脏利什曼病患者的63份临床样本(骨髓或脾脏抽吸物)。我们能够在几乎所有这些样品中(97%)使用一组诊断性SNP来鉴定基因型,并在大多数样品中(83%)获得全面的全基因组信息。这使我们能够进行系统遗传学分析,评估染色体拷贝数并鉴定大拷贝数变异体(CNV)。临床样本中的寄生虫基因组与衍生的体外培养的前鞭毛体之间的成对比较显示,变形虫的非整倍性较低,基因组差异也较小,表明患者存在多克隆感染。总而言之,我们的结果强调了直接在宿主样品中对寄生虫基因组进行测序的必要性。评估染色体拷贝数并确定大拷贝数变异体(CNV)。临床样本中的寄生虫基因组与衍生的体外培养的前鞭毛体之间的成对比较显示,变形虫的非整倍性较低,基因组差异也较小,表明患者存在多克隆感染。总而言之,我们的结果强调了直接在宿主样品中对寄生虫基因组进行测序的必要性。评估染色体拷贝数并确定大拷贝数变异体(CNV)。临床样本中的寄生虫基因组与衍生的体外培养的前鞭毛体之间的成对比较显示,变形虫的非整倍性较低,基因组差异也较小,表明患者存在多克隆感染。总而言之,我们的结果强调了直接在宿主样品中对寄生虫基因组进行测序的必要性。
更新日期:2019-12-13
中文翻译:
直接从印度次大陆患有内脏利什曼病的患者中测序利什曼原虫寄生虫的基因组。
全基因组测序(WGS)越来越多地用于传染病的分子诊断和流行病学。当前的利什曼原虫基因组研究依赖于从培养的寄生虫中提取的DNA,这可能会在随后的分析中引入采样和生物学偏见。到目前为止,临床样品中利什曼原虫基因组的直接分析受到人类DNA含量高和临床样品中寄生虫载量变化大的困扰。在这里,我们介绍一种基于具有安捷伦SureSelect技术的多形利什曼原虫DNA靶标富集的方法,该方法可直接在临床样品中分析利什曼原虫基因组。我们使用一组人工混合的样本验证了我们的方案,然后分析了尼泊尔内脏利什曼病患者的63份临床样本(骨髓或脾脏抽吸物)。我们能够在几乎所有这些样品中(97%)使用一组诊断性SNP来鉴定基因型,并在大多数样品中(83%)获得全面的全基因组信息。这使我们能够进行系统遗传学分析,评估染色体拷贝数并鉴定大拷贝数变异体(CNV)。临床样本中的寄生虫基因组与衍生的体外培养的前鞭毛体之间的成对比较显示,变形虫的非整倍性较低,基因组差异也较小,表明患者存在多克隆感染。总而言之,我们的结果强调了直接在宿主样品中对寄生虫基因组进行测序的必要性。评估染色体拷贝数并确定大拷贝数变异体(CNV)。临床样本中的寄生虫基因组与衍生的体外培养的前鞭毛体之间的成对比较显示,变形虫的非整倍性较低,基因组差异也较小,表明患者存在多克隆感染。总而言之,我们的结果强调了直接在宿主样品中对寄生虫基因组进行测序的必要性。评估染色体拷贝数并确定大拷贝数变异体(CNV)。临床样本中的寄生虫基因组与衍生的体外培养的前鞭毛体之间的成对比较显示,变形虫的非整倍性较低,基因组差异也较小,表明患者存在多克隆感染。总而言之,我们的结果强调了直接在宿主样品中对寄生虫基因组进行测序的必要性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号