当前位置:
X-MOL 学术
›
BBA Mol. Cell Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
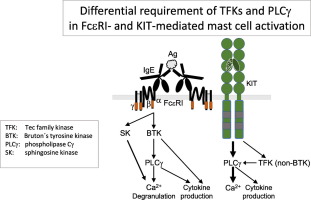
Differential use of BTK and PLC in FcεRI- and KIT-mediated mast cell activation: A marginal role of BTK upon KIT activation.
Biochimica et Biophysica Acta (BBA) - Molecular Cell Research ( IF 5.1 ) Pub Date : 2019-12-11 , DOI: 10.1016/j.bbamcr.2019.118622 Anne Simonowski 1 , Thomas Wilhelm 1 , Pardes Habib 2 , Carolin N Zorn 1 , Michael Huber 1
Biochimica et Biophysica Acta (BBA) - Molecular Cell Research ( IF 5.1 ) Pub Date : 2019-12-11 , DOI: 10.1016/j.bbamcr.2019.118622 Anne Simonowski 1 , Thomas Wilhelm 1 , Pardes Habib 2 , Carolin N Zorn 1 , Michael Huber 1
Affiliation
|
In mast cells (MCs), the TEC family kinase (TFK) BTK constitutes a central regulator of antigen (Ag)-triggered, FcεRI-mediated PLCγ phosphorylation, Ca2+ mobilization, degranulation, and pro-inflammatory cytokine production. Less is known about the function of BTK in the context of stem cell factor (SCF)-induced KIT signaling. In bone marrow-derived MCs (BMMCs), Ag stimulation caused intense phosphorylation of BTK at Y551 in its active center and at Y223 in its SH3-domain, whereas in response to SCF only Y223 was significantly phosphorylated. Further data using the TFK inhibitor Ibrutinib indicated that BTK Y223 is phosphorylated by a non-BTK TFK upon SCF stimulation. In line, SCF-induced PLCγ1 phosphorylation was stronger attenuated by Ibrutinib than by BTK deficiency. Subsequent pharmacological analysis of PLCγ function revealed a total block of SCF-induced Ca2+ mobilization by PLC inhibition, whereas only the sustained phase of Ca2+ flux was curtailed in Ag-stimulated BMMCs. Despite this severe stimulus-dependent difference in inducing Ca2+ mobilization, PLCγ inhibition suppressed Ag- and SCF-induced degranulation and pro-inflammatory cytokine production to comparable extents, suggesting involvement of additional TFK(s) or PLCγ-dependent signaling components. In addition to PLCγ, the MAPKs p38 and JNK were activated by Ag in a BTK-dependent manner; this was not observed upon SCF stimulation. Hence, FcεRI and KIT employ different mechanisms for activating PLCγ, p38, and JNK, which might strengthen their cooperation regarding pro-inflammatory MC effector functions. Importantly, our data clearly demonstrate that analyzing BTK Y223 phosphorylation is not sufficient to prove BTK activation.
中文翻译:

BTK和PLC在FcεRI和KIT介导的肥大细胞激活中的区别使用:BTK在KIT激活时的边际作用。
在肥大细胞(MC)中,TEC家族激酶(TFK)BTK构成了抗原(Ag)触发的,FcεRI介导的PLCγ磷酸化,Ca2 +动员,脱粒和促炎性细胞因子产生的中央调节剂。关于BTK在干细胞因子(SCF)诱导的KIT信号传导中的功能知之甚少。在骨髓衍生的MCs(BMMCs)中,Ag刺激在其活性中心的Y551和其SH3结构域的Y223处引起BTK的强烈磷酸化,而仅响应于SCF,Y223被显着磷酸化。使用TFK抑制剂依鲁替尼的进一步数据表明,在SCF刺激下,BTK Y223被非BTK TFK磷酸化。与之相比,依鲁替尼对SCF诱导的PLCγ1磷酸化的抑制作用比对BTK缺乏的抑制作用更强。随后对PLCγ功能的药理分析显示,PLC抑制可完全阻止SCF诱导的Ca2 +动员,而在Ag刺激的BMMC中仅Ca2 +通量的持续阶段被削减。尽管在诱导Ca2 +动员方面存在严重的依赖刺激的差异,但PLCγ抑制在相当程度上抑制了Ag和SCF诱导的脱粒和促炎性细胞因子的产生,这表明还涉及了其他TFK或PLCγ依赖的信号传导成分。除PLCγ外,Ag还以BTK依赖的方式激活MAPKs p38和JNK。在SCF刺激下未观察到此现象。因此,FcεRI和KIT采用不同的机制激活PLCγ,p38和JNK,这可能会增强它们在促炎性MC效应子功能方面的合作。重要的,
更新日期:2019-12-11
中文翻译:
BTK和PLC在FcεRI和KIT介导的肥大细胞激活中的区别使用:BTK在KIT激活时的边际作用。
在肥大细胞(MC)中,TEC家族激酶(TFK)BTK构成了抗原(Ag)触发的,FcεRI介导的PLCγ磷酸化,Ca2 +动员,脱粒和促炎性细胞因子产生的中央调节剂。关于BTK在干细胞因子(SCF)诱导的KIT信号传导中的功能知之甚少。在骨髓衍生的MCs(BMMCs)中,Ag刺激在其活性中心的Y551和其SH3结构域的Y223处引起BTK的强烈磷酸化,而仅响应于SCF,Y223被显着磷酸化。使用TFK抑制剂依鲁替尼的进一步数据表明,在SCF刺激下,BTK Y223被非BTK TFK磷酸化。与之相比,依鲁替尼对SCF诱导的PLCγ1磷酸化的抑制作用比对BTK缺乏的抑制作用更强。随后对PLCγ功能的药理分析显示,PLC抑制可完全阻止SCF诱导的Ca2 +动员,而在Ag刺激的BMMC中仅Ca2 +通量的持续阶段被削减。尽管在诱导Ca2 +动员方面存在严重的依赖刺激的差异,但PLCγ抑制在相当程度上抑制了Ag和SCF诱导的脱粒和促炎性细胞因子的产生,这表明还涉及了其他TFK或PLCγ依赖的信号传导成分。除PLCγ外,Ag还以BTK依赖的方式激活MAPKs p38和JNK。在SCF刺激下未观察到此现象。因此,FcεRI和KIT采用不同的机制激活PLCγ,p38和JNK,这可能会增强它们在促炎性MC效应子功能方面的合作。重要的,

























 京公网安备 11010802027423号
京公网安备 11010802027423号