当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Model systems for screening and investigation of lithium metal electrode chemistry and dendrite formation.
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2019-12-17 , DOI: 10.1039/c9cp06020k Ethan P Kamphaus 1 , Karoline Hight , Micah Dermott , Perla B Balbuena
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2019-12-17 , DOI: 10.1039/c9cp06020k Ethan P Kamphaus 1 , Karoline Hight , Micah Dermott , Perla B Balbuena
Affiliation
|
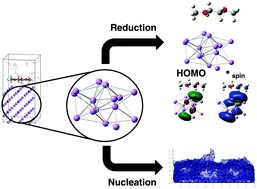
The use of lithium metal as an electrode for electrochemical energy storage will provide a significant impact on practical energy storage technology. Unfortunately, the use of lithium metal is plagued with challenging chemical problems. Specifically, the formation of a solid electrolyte interphase layer and the nucleation and growth of lithium dendrites: both must be addressed and controlled in order to achieve a practically useable pure lithium metal electrode. Currently sophisticated experimental techniques and computationally expensive simulations are being used to probe these problems but these methods are arduous and time consuming which delays timely evaluation and insight into the rapidly changing field of advanced energy storage. Here, we report the use of DFT simulations of lithium nanoclusters to investigate and explore lithium metal chemistry with inexpensive computational methods to gain greater insight into electrochemical reductions and the nucleation and growth of dendrites. DME, LiTFSI, and LiFSI reduction energetics and structures with electrode effects from lithium metal are reported providing better physical description of the absolute reduction potential characterization. The electronic structure of the lithium nanoclusters were used to investigate the nucleation and growth of lithium dendrites from an ab initio perspective. The results demonstrate that kinetic processes have more control over non uniform growth than thermodynamic processes. Based on this information, a non ab initio model was created in Matlab that shows the initial stages of dendrite nucleation considering approximately 2000 atoms.
中文翻译:

用于筛选和研究锂金属电极化学成分和枝晶形成的模型系统。
使用锂金属作为电化学能量存储的电极将对实际的能量存储技术产生重大影响。不幸的是,锂金属的使用困扰着具有挑战性的化学问题。具体而言,固体电解质中间层的形成以及锂树枝状晶体的成核和生长:必须同时解决和控制两者,以实现实用的纯锂金属电极。当前,正在使用复杂的实验技术和计算上昂贵的模拟来解决这些问题,但是这些方法艰巨且耗时,这延迟了及时评估和深入了解快速变化的先进储能领域的速度。这里,我们报告了使用DFT模拟锂纳米团簇,以廉价的计算方法来研究和探索锂金属化学,从而对电化学还原以及树枝状晶体的成核和生长有更深入的了解。据报道,DME,LiTFSI和LiFSI还原能量和具有锂金属电极效应的结构为绝对还原电位表征提供了更好的物理描述。锂纳米簇的电子结构用于从头开始研究锂树枝状晶体的形核和生长。结果表明,与热力学过程相比,动力学过程对非均匀生长的控制更多。根据此信息,
更新日期:2020-01-07
中文翻译:
用于筛选和研究锂金属电极化学成分和枝晶形成的模型系统。
使用锂金属作为电化学能量存储的电极将对实际的能量存储技术产生重大影响。不幸的是,锂金属的使用困扰着具有挑战性的化学问题。具体而言,固体电解质中间层的形成以及锂树枝状晶体的成核和生长:必须同时解决和控制两者,以实现实用的纯锂金属电极。当前,正在使用复杂的实验技术和计算上昂贵的模拟来解决这些问题,但是这些方法艰巨且耗时,这延迟了及时评估和深入了解快速变化的先进储能领域的速度。这里,我们报告了使用DFT模拟锂纳米团簇,以廉价的计算方法来研究和探索锂金属化学,从而对电化学还原以及树枝状晶体的成核和生长有更深入的了解。据报道,DME,LiTFSI和LiFSI还原能量和具有锂金属电极效应的结构为绝对还原电位表征提供了更好的物理描述。锂纳米簇的电子结构用于从头开始研究锂树枝状晶体的形核和生长。结果表明,与热力学过程相比,动力学过程对非均匀生长的控制更多。根据此信息,

























 京公网安备 11010802027423号
京公网安备 11010802027423号