当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Complete OSV-MP2 Analytical Gradient Theory for Molecular Structure and Dynamics Simulations.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2019-12-20 , DOI: 10.1021/acs.jctc.9b00806 Ruiyi Zhou 1 , Qiujiang Liang 1 , Jun Yang 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2019-12-20 , DOI: 10.1021/acs.jctc.9b00806 Ruiyi Zhou 1 , Qiujiang Liang 1 , Jun Yang 1
Affiliation
|
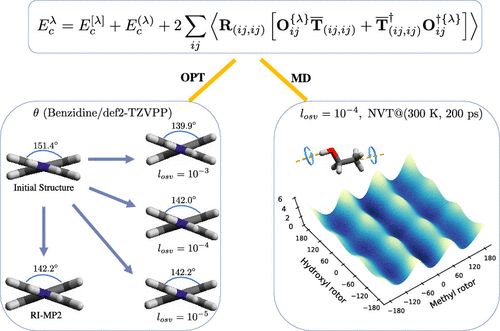
We propose an exact algorithm for computing the analytical gradient within the framework of the orbital-specific-virtual (OSV) second-order Møller-Plesset (MP2) theory in resolution-of-identity (RI) approximation. We implement the relaxation of perturbed OSVs through the explicit constraints of the perturbed orthonormality, the perturbed diagonality, and the perturbed eigenvalue condition. We show that the rotation of OSVs within the retained OSV subspace makes no contribution to gradients, as long as the unperturbed Hylleraas energy functional reaches minimum. The OSV relaxation is solved as the perturbed nondegenerate eigenvalue problem between the retained and discarded OSV subspaces. The detailed derivation and preliminary implementations for gradient working equations are discussed. The coupled-perturbed localization method is implemented for meta-Löwdin localization function. The numerical accuracy of computed OSV-MP2 gradients is demonstrated for the geometries of selected molecules that are often discussed in other theories. From OSV-MP2 with the normal OSV selection, the canonical RI-MP2/def2-TZVP gradients can be reproduced within 10-4 au. The OSV-MP2/def2-TZVPP covalent bond lengths, angles, and dihedral angles are in good agreement with canonical RI-MP2 structures by 0.017 pm, 0.03°, and 0.2°, respectively. No particular accuracy gains have been observed for molecular geometries compared to the recent local pair-natural-orbital MP2 by using the predefined orbital domains. Moreover, the OSV-MP2 analytical gradients can generate atomic forces that are utilized to drive the Born-Oppenheimer molecular dynamics (BOMD) simulation for studying structural and vibrational properties with respect to OSV selections. By performing the OSV-MP2 NVE BOMD calculation using the normal OSV selection, the structural and vibrational details of protonated water cations are well reproduced. The 200 ps NVT well-tempered metadynamics at 300 K has been simulated to compute the OSV-MP2 rotational free-energy surface of coupled hydroxyl and methyl rotors for ethanol molecule.
中文翻译:

完整的OSV-MP2分析梯度理论,用于分子结构和动力学模拟。
我们提出了一种精确的算法,用于在恒等式(RI)逼近中的轨道特定虚拟(OSV)二阶Møller-Plesset(MP2)理论的框架内计算分析梯度。我们通过摄动正交正态,摄动对角线和摄动特征值条件的显式约束来实现摄动OSV的松弛。我们表明,只要不受扰动的Hylleraas能量函数达到最小值,在保留的OSV子空间内OSV的旋转就不会对梯度做出任何贡献。解决了OSV松弛问题,作为保留和丢弃的OSV子空间之间的扰动的非退化特征值问题。讨论了梯度工作方程的详细推导和初步实现。耦合摄动定位方法是针对meta-Löwdin定位功能实现的。对于其他理论中经常讨论的所选分子的几何结构,已证明了计算出的OSV-MP2梯度的数值准确性。从具有正常OSV选择的OSV-MP2,可以在10-4 au之内再现典型的RI-MP2 / def2-TZVP梯度。OSV-MP2 / def2-TZVPP共价键的长度,角度和二面角分别与规范的RI-MP2结构相符,分别为0.017 pm,0.03°和0.2°。通过使用预定义的轨道域,与最近的局部对-自然轨道MP2相比,没有观察到分子几何结构的特别准确度提高。而且,OSV-MP2的分析梯度可以生成原子力,该原子力用于驱动Born-Oppenheimer分子动力学(BOMD)模拟,以研究有关OSV选择的结构和振动特性。通过使用常规OSV选择执行OSV-MP2 NVE BOMD计算,可以很好地再现质子化水阳离子的结构和振动细节。已经模拟了在300 K时200 ps NVT充分调和的动力学,以计算乙醇分子的耦合的羟基和甲基转子的OSV-MP2旋转自由能表面。
更新日期:2019-12-21
中文翻译:
完整的OSV-MP2分析梯度理论,用于分子结构和动力学模拟。
我们提出了一种精确的算法,用于在恒等式(RI)逼近中的轨道特定虚拟(OSV)二阶Møller-Plesset(MP2)理论的框架内计算分析梯度。我们通过摄动正交正态,摄动对角线和摄动特征值条件的显式约束来实现摄动OSV的松弛。我们表明,只要不受扰动的Hylleraas能量函数达到最小值,在保留的OSV子空间内OSV的旋转就不会对梯度做出任何贡献。解决了OSV松弛问题,作为保留和丢弃的OSV子空间之间的扰动的非退化特征值问题。讨论了梯度工作方程的详细推导和初步实现。耦合摄动定位方法是针对meta-Löwdin定位功能实现的。对于其他理论中经常讨论的所选分子的几何结构,已证明了计算出的OSV-MP2梯度的数值准确性。从具有正常OSV选择的OSV-MP2,可以在10-4 au之内再现典型的RI-MP2 / def2-TZVP梯度。OSV-MP2 / def2-TZVPP共价键的长度,角度和二面角分别与规范的RI-MP2结构相符,分别为0.017 pm,0.03°和0.2°。通过使用预定义的轨道域,与最近的局部对-自然轨道MP2相比,没有观察到分子几何结构的特别准确度提高。而且,OSV-MP2的分析梯度可以生成原子力,该原子力用于驱动Born-Oppenheimer分子动力学(BOMD)模拟,以研究有关OSV选择的结构和振动特性。通过使用常规OSV选择执行OSV-MP2 NVE BOMD计算,可以很好地再现质子化水阳离子的结构和振动细节。已经模拟了在300 K时200 ps NVT充分调和的动力学,以计算乙醇分子的耦合的羟基和甲基转子的OSV-MP2旋转自由能表面。


























 京公网安备 11010802027423号
京公网安备 11010802027423号