当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Efficient Formulation of Ab Initio Quantum Embedding in Periodic Systems: Dynamical Mean-Field Theory.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-01-03 , DOI: 10.1021/acs.jctc.9b00934 Tianyu Zhu 1 , Zhi-Hao Cui 1 , Garnet Kin-Lic Chan 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-01-03 , DOI: 10.1021/acs.jctc.9b00934 Tianyu Zhu 1 , Zhi-Hao Cui 1 , Garnet Kin-Lic Chan 1
Affiliation
|
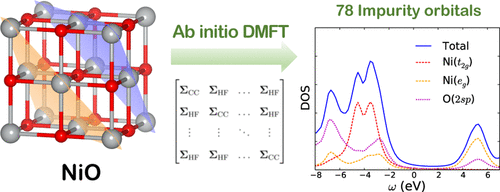
We present an efficient ab initio dynamical mean-field theory (DMFT) implementation for quantitative simulations in solids. Our DMFT scheme employs ab initio Hamiltonians defined for impurities comprising the full unit cell or a supercell of atoms and for realistic quantum chemical basis sets. We avoid double counting errors by using Hartree-Fock as the low-level theory. Intrinsic and projected atomic orbitals (IAO + PAO) are chosen as the local embedding basis, facilitating numerical bath truncation. Using an efficient integral transformation and coupled-cluster Green's function impurity solvers, we are able to handle embedded impurity problems with several hundred orbitals. We apply our ab initio DMFT approach to study a hexagonal boron nitride monolayer, crystalline silicon, and nickel oxide in the antiferromagnetic phase, with up to 104 and 78 impurity orbitals in the spin-restricted and unrestricted cluster DMFT calculations and over 100 bath orbitals. We show that our scheme produces accurate spectral functions compared to both benchmark periodic coupled-cluster computations and experimental spectra.
中文翻译:

周期系统中从头算起的量子嵌入的有效表示:动力学平均场理论。
我们提出了一种有效的从头算平均动力学理论(DMFT)的实现方法,用于固体中的定量模拟。我们的DMFT方案采用从头算起的哈密顿量,其定义为包含原子的完整晶胞或超晶胞的杂质以及现实的量子化学基础集。我们通过使用Hartree-Fock作为低层理论来避免重复计算错误。选择固有的和投影的原子轨道(IAO + PAO)作为局部嵌入基础,以简化数值浴槽截断。使用有效的积分变换和耦合簇格林函数杂质求解器,我们能够处理数百个轨道的嵌入式杂质问题。我们采用从头开始DMFT方法研究反铁磁相中的六方氮化硼单层,晶体硅和氧化镍,在自旋限制和无限制簇DMFT计算中,最多有104和78个杂质轨道,并且有超过100个浴槽轨道。我们表明,与基准周期耦合群集计算和实验光谱相比,我们的方案可产生准确的光谱函数。
更新日期:2020-01-04
中文翻译:
周期系统中从头算起的量子嵌入的有效表示:动力学平均场理论。
我们提出了一种有效的从头算平均动力学理论(DMFT)的实现方法,用于固体中的定量模拟。我们的DMFT方案采用从头算起的哈密顿量,其定义为包含原子的完整晶胞或超晶胞的杂质以及现实的量子化学基础集。我们通过使用Hartree-Fock作为低层理论来避免重复计算错误。选择固有的和投影的原子轨道(IAO + PAO)作为局部嵌入基础,以简化数值浴槽截断。使用有效的积分变换和耦合簇格林函数杂质求解器,我们能够处理数百个轨道的嵌入式杂质问题。我们采用从头开始DMFT方法研究反铁磁相中的六方氮化硼单层,晶体硅和氧化镍,在自旋限制和无限制簇DMFT计算中,最多有104和78个杂质轨道,并且有超过100个浴槽轨道。我们表明,与基准周期耦合群集计算和实验光谱相比,我们的方案可产生准确的光谱函数。


























 京公网安备 11010802027423号
京公网安备 11010802027423号