当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Hierarchical Ensembles of Intrinsically Disordered Proteins at Atomic Resolution in Molecular Dynamics Simulations.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00809 Lisa M Pietrek 1 , Lukas S Stelzl 1 , Gerhard Hummer 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00809 Lisa M Pietrek 1 , Lukas S Stelzl 1 , Gerhard Hummer 1, 2
Affiliation
|
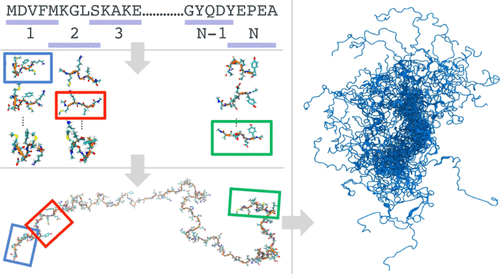
Intrinsically disordered proteins (IDPs) constitute a large fraction of the human proteome and are critical in the regulation of cellular processes. A detailed understanding of the conformational dynamics of IDPs could help to elucidate their roles in health and disease. However, the inherent flexibility of IDPs makes structural studies and their interpretation challenging. Molecular dynamics (MD) simulations could address this challenge in principle, but inaccuracies in the simulation models and the need for long simulations have stymied progress. To overcome these limitations, we adopt a hierarchical approach that builds on the "flexible-meccano" model reported by Bernadó et al. (J. Am. Chem. Soc. 2005, 127, 17968-17969). First, we exhaustively sample small IDP fragments in all-atom simulations to capture their local structures. Then, we assemble the fragments into full-length IDPs to explore the stereochemically possible global structures of IDPs. The resulting ensembles of three-dimensional structures of full-length IDPs are highly diverse, much more so than in standard MD simulation. For the paradigmatic IDP α-synuclein, our ensemble captures both the local structure, as probed by nuclear magnetic resonance spectroscopy, and its overall dimension, as obtained from small-angle X-ray scattering in solution. By generating representative and meaningful starting ensembles, we can begin to exploit the massive parallelism afforded by current and future high-performance computing resources for atomic-resolution characterization of IDPs.
中文翻译:

分子动力学模拟中原子分辨的固有无序蛋白质的层次集成。
本质上无序的蛋白质(IDP)构成了人类蛋白质组的很大一部分,并且在调节细胞过程中起着至关重要的作用。对IDP构象动力学的详细了解可以帮助阐明其在健康和疾病中的作用。但是,国内流离失所者固有的灵活性使结构研究及其解释具有挑战性。分子动力学(MD)模拟原则上可以解决这一难题,但是模拟模型的不精确性以及对长时间模拟的需求已阻碍了进展。为了克服这些限制,我们采用了一种基于Bernadó等人报道的“ flexible-meccano”模型的分层方法。(J.Am.Chem.Soc.2005,127,17968-17969)。首先,我们在全原子模拟中详尽地采样了小的IDP碎片,以捕获其局部结构。然后,我们将这些片段组装成全长IDP,以探索IDP的立体化学可能的整体结构。全长IDP的三维结构的结果集合非常多样化,比标准MD模拟中的结果要丰富得多。对于典型的IDPα-突触核蛋白,我们的集合既捕获了通过核磁共振波谱探测的局部结构,又捕获了从溶液中的小角度X射线散射获得的整体结构。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。全长IDP的三维结构的集成非常多样化,比标准的MD仿真要复杂得多。对于典型的IDPα-突触核蛋白,我们的集合既捕获了通过核磁共振波谱探测的局部结构,又捕获了从溶液中的小角度X射线散射获得的整体结构。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。全长IDP的三维结构的结果集合非常多样化,比标准MD模拟中的结果要丰富得多。对于典型的IDPα-突触核蛋白,我们的集合既捕获了通过核磁共振波谱探测的局部结构,又捕获了从溶液中的小角度X射线散射获得的整体结构。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。由溶液中的小角度X射线散射获得的图像及其整体尺寸。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。由溶液中的小角度X射线散射获得的图像及其整体尺寸。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。
更新日期:2019-12-27
中文翻译:
分子动力学模拟中原子分辨的固有无序蛋白质的层次集成。
本质上无序的蛋白质(IDP)构成了人类蛋白质组的很大一部分,并且在调节细胞过程中起着至关重要的作用。对IDP构象动力学的详细了解可以帮助阐明其在健康和疾病中的作用。但是,国内流离失所者固有的灵活性使结构研究及其解释具有挑战性。分子动力学(MD)模拟原则上可以解决这一难题,但是模拟模型的不精确性以及对长时间模拟的需求已阻碍了进展。为了克服这些限制,我们采用了一种基于Bernadó等人报道的“ flexible-meccano”模型的分层方法。(J.Am.Chem.Soc.2005,127,17968-17969)。首先,我们在全原子模拟中详尽地采样了小的IDP碎片,以捕获其局部结构。然后,我们将这些片段组装成全长IDP,以探索IDP的立体化学可能的整体结构。全长IDP的三维结构的结果集合非常多样化,比标准MD模拟中的结果要丰富得多。对于典型的IDPα-突触核蛋白,我们的集合既捕获了通过核磁共振波谱探测的局部结构,又捕获了从溶液中的小角度X射线散射获得的整体结构。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。全长IDP的三维结构的集成非常多样化,比标准的MD仿真要复杂得多。对于典型的IDPα-突触核蛋白,我们的集合既捕获了通过核磁共振波谱探测的局部结构,又捕获了从溶液中的小角度X射线散射获得的整体结构。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。全长IDP的三维结构的结果集合非常多样化,比标准MD模拟中的结果要丰富得多。对于典型的IDPα-突触核蛋白,我们的集合既捕获了通过核磁共振波谱探测的局部结构,又捕获了从溶液中的小角度X射线散射获得的整体结构。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。由溶液中的小角度X射线散射获得的图像及其整体尺寸。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。由溶液中的小角度X射线散射获得的图像及其整体尺寸。通过生成有代表性的有意义的初始集合,我们可以开始利用当前和将来的高性能计算资源提供的大规模并行性来对IDP进行原子分辨率表征。


























 京公网安备 11010802027423号
京公网安备 11010802027423号