当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Comparison of Statistical Tests and Power Analysis for Phosphoproteomics Data.
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jproteome.9b00280 Lei J Ding , Hannah M Schlüter 1 , Matthew J Szucs 2 , Rushdy Ahmad 2 , Zheyang Wu 3 , Weifeng Xu
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jproteome.9b00280 Lei J Ding , Hannah M Schlüter 1 , Matthew J Szucs 2 , Rushdy Ahmad 2 , Zheyang Wu 3 , Weifeng Xu
Affiliation
|
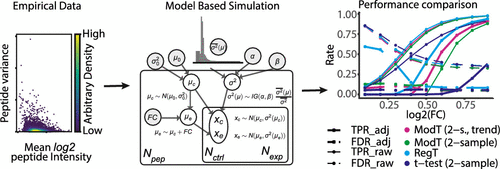
Advances in protein tagging and mass spectrometry have enabled generation of large quantitative proteome and phosphoproteome data sets, for identifying differentially expressed targets in case-control studies. The power study of statistical tests is critical for designing strategies for effective target identification and control of experimental cost. Here, we develop a simulation framework to generate realistic phospho-peptide data with known changes between cases and controls. Using this framework, we quantify the performance of traditional t-tests, Bayesian tests, and the ranking-by-fold-change test. Bayesian tests, which share variance information among peptides, outperform the traditional t-tests. Although ranking-by-fold-change has similar power as the Bayesian tests, its type I error rate cannot be properly controlled without proper permutation analysis; therefore, simply relying on the ranking likely brings false positives. Two-sample Bayesian tests considering dependencies between intensity and variance are superior for data sets with complex variance. While increasing the sample size enhances the statistical tests' performance, balanced controls and cases are recommended over a one-side weighted group. Further, higher peptide standard deviations require higher fold changes to achieve the same statistical power. Together, these results highlight the importance of model-informed experimental design and principled statistical analyses when working with large-scale proteomics and phosphoproteomics data.
中文翻译:

磷酸蛋白质组学数据的统计测试和功效分析的比较。
蛋白质标签和质谱技术的进步使得能够生成大量的定量蛋白质组和磷酸化蛋白质组数据集,以便在病例对照研究中鉴定差异表达的靶标。统计测试的能力研究对于设计有效目标识别和控制实验成本的策略至关重要。在这里,我们开发了一个模拟框架,以生成实际的磷酸肽数据,并在病例和对照之间进行已知的更改。使用此框架,我们可以量化传统t检验,贝叶斯检验和按变化倍数排序检验的性能。在肽段之间共享方差信息的贝叶斯测试优于传统的t检验。尽管按变化排位具有与贝叶斯测试相似的功能,如果没有适当的置换分析,则无法正确控制其I型错误率;因此,仅依靠排名可能会带来误报。考虑到强度和方差之间的相关性的两样本贝叶斯检验对于具有复杂方差的数据集是优越的。虽然增加样本量可增强统计检验的性能,但建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在处理大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。考虑到强度和方差之间的相关性的两样本贝叶斯检验对于具有复杂方差的数据集是优越的。虽然增加样本量可增强统计检验的性能,但建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。考虑到强度和方差之间的相关性的两样本贝叶斯检验对于具有复杂方差的数据集是优越的。虽然增加样本量可增强统计检验的性能,但建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。建议在一侧加权组中保持平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。
更新日期:2019-12-27
中文翻译:
磷酸蛋白质组学数据的统计测试和功效分析的比较。
蛋白质标签和质谱技术的进步使得能够生成大量的定量蛋白质组和磷酸化蛋白质组数据集,以便在病例对照研究中鉴定差异表达的靶标。统计测试的能力研究对于设计有效目标识别和控制实验成本的策略至关重要。在这里,我们开发了一个模拟框架,以生成实际的磷酸肽数据,并在病例和对照之间进行已知的更改。使用此框架,我们可以量化传统t检验,贝叶斯检验和按变化倍数排序检验的性能。在肽段之间共享方差信息的贝叶斯测试优于传统的t检验。尽管按变化排位具有与贝叶斯测试相似的功能,如果没有适当的置换分析,则无法正确控制其I型错误率;因此,仅依靠排名可能会带来误报。考虑到强度和方差之间的相关性的两样本贝叶斯检验对于具有复杂方差的数据集是优越的。虽然增加样本量可增强统计检验的性能,但建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在处理大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。考虑到强度和方差之间的相关性的两样本贝叶斯检验对于具有复杂方差的数据集是优越的。虽然增加样本量可增强统计检验的性能,但建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。考虑到强度和方差之间的相关性的两样本贝叶斯检验对于具有复杂方差的数据集是优越的。虽然增加样本量可增强统计检验的性能,但建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。建议在一侧加权组中使用平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。建议在一侧加权组中保持平衡的对照和病例。此外,较高的肽标准偏差需要较高的倍数变化才能达到相同的统计功效。总之,这些结果凸显了在使用大规模蛋白质组学和磷酸化蛋白质组学数据时,进行模型研究的实验设计和原则化的统计分析的重要性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号