JAMA Oncology ( IF 28.4 ) Pub Date : 2020-01-01 , DOI: 10.1001/jamaoncol.2019.3985 Alexander Penson 1, 2, 3 , Niedzica Camacho 1, 2, 4 , Youyun Zheng 2, 4 , Anna M Varghese 5 , Hikmat Al-Ahmadie 4 , Pedram Razavi 5 , Sarat Chandarlapaty 1, 5 , Christina E Vallejo 4 , Efsevia Vakiani 2, 4 , Teresa Gilewski 5 , Jonathan E Rosenberg 5 , Maha Shady 2, 4 , Dana W Y Tsui 2, 4 , Dalicia N Reales 6 , Adam Abeshouse 1, 2, 3 , Aijazuddin Syed 4 , Ahmet Zehir 4 , Nikolaus Schultz 1, 2, 3 , Marc Ladanyi 1, 4 , David B Solit 1, 2, 5, 7 , David S Klimstra 4, 8 , David M Hyman 5, 7 , Barry S Taylor 1, 2, 3 , Michael F Berger 1, 2, 4, 8
|
Importance Diagnosing the site of origin for cancer is a pillar of disease classification that has directed clinical care for more than a century. Even in an era of precision oncologic practice, in which treatment is increasingly informed by the presence or absence of mutant genes responsible for cancer growth and progression, tumor origin remains a critical factor in tumor biologic characteristics and therapeutic sensitivity.
Objective To evaluate whether data derived from routine clinical DNA sequencing of tumors could complement conventional approaches to enable improved diagnostic accuracy.
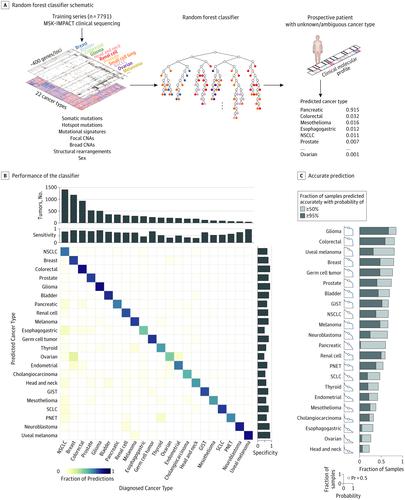
Design, Setting, and Participants A machine learning approach was developed to predict tumor type from targeted panel DNA sequence data obtained at the point of care, incorporating both discrete molecular alterations and inferred features such as mutational signatures. This algorithm was trained on 7791 tumors representing 22 cancer types selected from a prospectively sequenced cohort of patients with advanced cancer.
Results The correct tumor type was predicted for 5748 of the 7791 patients (73.8%) in the training set as well as 8623 of 11 644 patients (74.1%) in an independent cohort. Predictions were assigned probabilities that reflected empirical accuracy, with 3388 cases (43.5%) representing high-confidence predictions (>95% probability). Informative molecular features and feature categories varied widely by tumor type. Genomic analysis of plasma cell-free DNA yielded accurate predictions in 45 of 60 cases (75.0%), suggesting that this approach may be applied in diverse clinical settings including as an adjunct to cancer screening. Likely tissues of origin were predicted from targeted tumor sequencing in 95 of 141 patients (67.4%) with cancers of unknown primary site. Applying this method prospectively to patients under active care enabled genome-directed reassessment of diagnosis in 2 patients initially presumed to have metastatic breast cancer, leading to the selection of more appropriate treatments, which elicited clinical responses.
Conclusions and Relevance These results suggest that the application of artificial intelligence to predict tissue of origin in oncologic practice can act as a useful complement to conventional histologic review to provide integrated pathologic diagnoses, often with important therapeutic implications.
中文翻译:
基因组衍生肿瘤类型预测的发展为临床癌症护理提供信息
重要性 诊断癌症的起源部位是疾病分类的一个支柱,一个多世纪以来一直指导着临床护理。即使在精准肿瘤学实践的时代,治疗越来越多地取决于是否存在负责癌症生长和进展的突变基因,肿瘤起源仍然是肿瘤生物学特征和治疗敏感性的关键因素。
目的 评估来自肿瘤常规临床 DNA 测序的数据是否可以补充传统方法,以提高诊断准确性。
设计、设置和参与者 开发了一种机器学习方法,用于根据护理时获得的目标组 DNA 序列数据预测肿瘤类型,结合离散分子改变和推断特征(如突变特征)。该算法针对代表 22 种癌症类型的 7791 个肿瘤进行了训练,这些肿瘤选自前瞻性测序的晚期癌症患者队列。
结果 训练 集中 7791 名患者中的 5748 名患者 (73.8%) 以及独立队列中 11 644 名患者中的 8623 名患者 (74.1%) 预测了正确的肿瘤类型。预测被分配了反映经验准确性的概率,其中 3388 个案例 (43.5%) 代表高置信度预测(>95% 的概率)。信息分子特征和特征类别因肿瘤类型而异。血浆游离 DNA 的基因组分析在 60 例病例中的 45 例 (75.0%) 中产生了准确的预测,表明这种方法可应用于多种临床环境,包括作为癌症筛查的辅助手段。通过靶向肿瘤测序,对 141 名原发部位不明的癌症患者中的 95 名 (67.4%) 进行了预测,预测了可能的起源组织。将这种方法前瞻性地应用于接受积极护理的患者,能够对两名最初被认为患有转移性乳腺癌的患者进行基因组定向重新评估,从而选择更合适的治疗方法,从而引起临床反应。
结论和相关性 这些结果表明,在肿瘤学实践中应用人工智能来预测组织起源可以作为传统组织学检查的有用补充,以提供综合病理诊断,通常具有重要的治疗意义。


























 京公网安备 11010802027423号
京公网安备 11010802027423号