Journal of Biomedical informatics ( IF 4.5 ) Pub Date : 2019-10-23 , DOI: 10.1016/j.jbi.2019.103313 Md Ali Hossain 1 , Sheikh Muhammad Saiful Islam 2 , Julian M W Quinn 3 , Fazlul Huq 4 , Mohammad Ali Moni 5
|
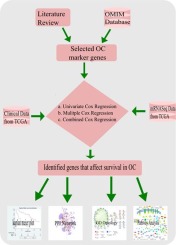
Ovarian cancer (OC) is a common cause of cancer death among women worldwide, so there is a pressing need to identify factors influencing OC mortality. Much OC patient clinical data is publicly accessible via the Broad Institute Cancer Genome Atlas (TCGA) datasets which include patient age, cancer site, stage and subtype and patient survival, as well as OC gene transcription profiles. These allow studies correlating OC patient survival (and other clinical variables) with gene expression to identify new OC biomarkers to predict patient mortality. We integrated clinical and tissue transcriptome data from patients available from the TCGA portal. We determined OC mRNA expression levels (compared to normal ovarian tissue) of 41 genes already implicated in OC progression, and assessed how their OC tissue expression levels predicts patient survival. We employed Cox Proportional Hazard regression models to analyse clinical factors and transcriptomic information to determine the relative effects on survival that is associated with each factor. Multivariate analysis of combined data (clinical and gene mRNA expression) found age and ovary tumour site significantly correlated with patient survival. The univariate analysis also confirmed significant differences in patient survival time when altered transcription levels of TLR4, BSCL2, CDH1, ERBB2, and SCGB2A1 were evident, while multivariate analysis that considered the 41 genes simultaneously revealed a significant relationship of survival with TLR4, BSCL2, CDH1, ERBB2 and PTPRE genes. However, analyses that considered all 41 genes with clinical variables together identified genes TLR4, BSCL2, CDH1, ERBB2, BRCA2 and SCGB2A1 as independently related to survival in OC. These studies indicate that the latter genes influence OC patient survival, i.e., expression levels of these genes provide mechanistic and predictive information in addition to that of the clinical traits. Our study provides strong evidence that these genes are important prognostic indicators of patient survival that give clues to biological processes that underlie OC progression and mortality.
中文翻译:
机器学习和生物信息学模型可识别与疾病进展和死亡率相关的卵巢癌基因表达模式。
卵巢癌(OC)是全世界女性癌症死亡的常见原因,因此迫切需要确定影响OC死亡率的因素。可通过Broad Institute Cancer Genome Atlas(TCGA)数据集公开获得许多OC患者的临床数据,其中包括患者年龄,癌症部位,分期和亚型以及患者生存率以及OC基因转录谱。这些研究使OC患者的生存(和其他临床变量)与基因表达相关联,从而鉴定出新的OC生物标志物来预测患者的死亡率。我们整合了来自TCGA门户网站的患者的临床和组织转录组数据。我们确定了已经与OC进展相关的41个基因的OC mRNA表达水平(与正常卵巢组织相比),并评估了它们的OC组织表达水平如何预测患者的存活率。我们采用Cox比例危害回归模型分析临床因素和转录组信息,以确定与每个因素相关的生存率的相对影响。综合数据(临床和基因mRNA表达)的多变量分析发现,年龄和卵巢肿瘤部位与患者生存率显着相关。当TLR4,BSCL2,CDH1,ERBB2和SCGB2A1的转录水平发生改变时,单变量分析还证实了患者生存时间的显着差异,而同时考虑了41个基因的多变量分析则显示了生存与TLR4,BSCL2,CDH1的显着相关性,ERBB2和PTPRE基因。但是,经过综合考虑所有41个具有临床变量的基因的分析共同确定了TLR4,BSCL2,CDH1,ERBB2,BRCA2和SCGB2A1与OC生存独立相关。这些研究表明,后者的基因会影响OC患者的生存,即这些基因的表达水平除了提供临床特征外,还提供了机械和预测性信息。我们的研究提供了有力的证据,表明这些基因是患者生存的重要预后指标,为提示OC进展和死亡率的生物学过程提供了线索。
























 京公网安备 11010802027423号
京公网安备 11010802027423号