当前位置:
X-MOL 学术
›
J. Chin. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Experimental and theoretical electronic absorption spectra of 2,4‐diphenyl‐1,5‐benzothiazepine and its derivatives: Solvatochromic effect and time dependent density functional theory approach
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2019-07-21 , DOI: 10.1002/jccs.201900063 Hussein Moustafa 1 , Mohamed E. Elshakre 1 , Huwaida M. E. Hassaneen 1 , Salwa Elramly 1
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2019-07-21 , DOI: 10.1002/jccs.201900063 Hussein Moustafa 1 , Mohamed E. Elshakre 1 , Huwaida M. E. Hassaneen 1 , Salwa Elramly 1
Affiliation
|
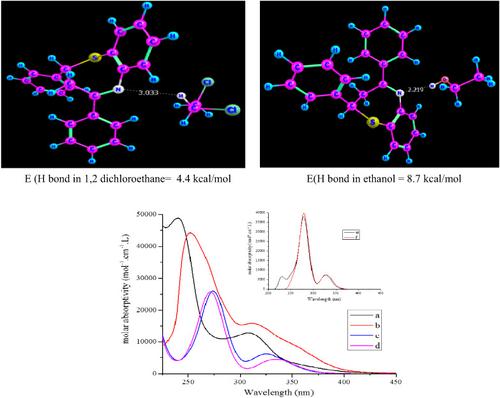
Electronic spectra of 2,4‐diphenyl‐1,5‐benzothiazepine and some of its derivatives in 1,2‐dichloromethane and ethanol are investigated experimentally and theoretically using the time dependent density functional theory (TD‐DFT) method at the B3LYP/6‐311G** level of the theory. The origin of the spectrum of the parent compound is found to be an additive one. The observed ultra violet (UV) spectra in both solvents show two bands S1 in the range between 312–334 nm and S2 in the range between 248–272 nm. The solvent effect is investigated experimentally and theoretically and a blue shift is observed, which is explained in terms of a hydrogen bond model between the solvent and the most negative site of the solute (N atom). This theoretical model is robust in reproducing the experimental blue shift and calculating the hydrogen bond energy and hydrogen bond length. The extent of delocalization and charge transfer processes of the studied compounds is estimated and discussed in terms of natural bond orbital (NBO) analysis and second order perturbation interactions (E2) between donors and acceptors. The effect of substituents of the studied compounds in both solvents shows a noticeable red shift attributed to hyperconjugation effects of the π electron systems of the different moieties.
中文翻译:

2,4-二苯基-1,5-苯并噻嗪类及其衍生物的实验和理论电子吸收光谱:溶剂变色效应和时变密度泛函理论方法
在B3LYP / 6上使用时变密度泛函理论(TD-DFT)方法对理论上的理论和实验方法研究了2,4-二苯基-1,5-苯并噻吩并在1,2-二氯甲烷和乙醇中的某些电子光谱‐311G **理论水平。发现母体化合物的光谱的起源是加和的。在两种溶剂中观察到的紫外(UV)光谱均显示在312–334 nm范围内的两个谱带S1和在248–272 nm范围内的S2。通过实验和理论研究了溶剂效应,并观察到蓝移,这是通过溶剂与溶质(N原子)最负部位之间的氢键模型来解释的。该理论模型在再现实验蓝移和计算氢键能和氢键长度方面具有鲁棒性。根据自然键轨道(NBO)分析和二阶扰动相互作用,估算并讨论了所研究化合物的离域和电荷转移过程的程度(E 2)在捐赠者和接受者之间。研究的化合物在两种溶剂中的取代基作用均显示出明显的红移,这归因于不同部分的π电子系统的超共轭作用。
更新日期:2019-12-11
中文翻译:
2,4-二苯基-1,5-苯并噻嗪类及其衍生物的实验和理论电子吸收光谱:溶剂变色效应和时变密度泛函理论方法
在B3LYP / 6上使用时变密度泛函理论(TD-DFT)方法对理论上的理论和实验方法研究了2,4-二苯基-1,5-苯并噻吩并在1,2-二氯甲烷和乙醇中的某些电子光谱‐311G **理论水平。发现母体化合物的光谱的起源是加和的。在两种溶剂中观察到的紫外(UV)光谱均显示在312–334 nm范围内的两个谱带S1和在248–272 nm范围内的S2。通过实验和理论研究了溶剂效应,并观察到蓝移,这是通过溶剂与溶质(N原子)最负部位之间的氢键模型来解释的。该理论模型在再现实验蓝移和计算氢键能和氢键长度方面具有鲁棒性。根据自然键轨道(NBO)分析和二阶扰动相互作用,估算并讨论了所研究化合物的离域和电荷转移过程的程度(E 2)在捐赠者和接受者之间。研究的化合物在两种溶剂中的取代基作用均显示出明显的红移,这归因于不同部分的π电子系统的超共轭作用。

























 京公网安备 11010802027423号
京公网安备 11010802027423号