当前位置:
X-MOL 学术
›
BBA Proteins Proteom.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Quantitative proteomics indicate a strong correlation of mitotic phospho-/dephosphorylation with non-structured regions of substrates.
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics ( IF 3.2 ) Pub Date : 2019-10-30 , DOI: 10.1016/j.bbapap.2019.140295 Hiroya Yamazaki 1 , Hidetaka Kosako 2 , Shige H Yoshimura 1
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics ( IF 3.2 ) Pub Date : 2019-10-30 , DOI: 10.1016/j.bbapap.2019.140295 Hiroya Yamazaki 1 , Hidetaka Kosako 2 , Shige H Yoshimura 1
Affiliation
|
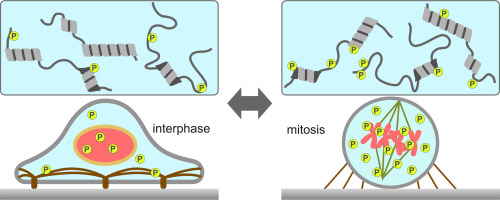
Protein phosphorylation plays a critical role in the regulation and progression of mitosis. >40,000 phosphorylated residues and the associated kinases have been identified to date via proteomic analyses. Although some of these phosphosites are associated with regulation of either protein-protein interactions or the catalytic activity of the substrate protein, the roles of most mitotic phosphosites remain unclear. In this study, we examined structural properties of mitotic phosphosites and neighboring residues to understand the role of heavy phosphorylation in non-structured domains. Quantitative mass spectrometry analysis of mitosis-arrested and non-arrested HeLa cells revealed >4100 and > 2200 residues either significantly phosphorylated or dephosphorylated, respectively, at mitotic entry. The calculated disorder scores of amino acid sequences of neighboring individual phosphosites revealed that >70% of dephosphorylated phosphosites exist in disordered regions, whereas 50% of phosphorylated sites exist in non-structured domains. A clear inverse correlation was observed between probability of phosphorylation in non-structured domain and increment of phosphorylation in mitosis. These results indicate that at entry to mitosis, a significant number of phosphate groups are removed from non-structured domains and transferred to more-structured domains. Gene ontology term analysis revealed that mitosis-related proteins are heavily phosphorylated, whereas RNA-related proteins are both dephosphorylated and phosphorylated, suggesting that heavy phosphorylation/dephosphorylation in non-structured domains of RNA-binding proteins plays a role in dynamic rearrangement of RNA-containing organelles, as well as other intracellular environments.
中文翻译:

定量蛋白质组学表明有丝分裂的磷酸化/去磷酸化与底物的非结构化区域有很强的相关性。
蛋白质磷酸化在有丝分裂的调控和进程中起着至关重要的作用。迄今为止,通过蛋白质组学分析已鉴定出> 40,000个磷酸化残基和相关的激酶。尽管这些磷酸酯中的一些与调节蛋白质-蛋白质相互作用或底物蛋白的催化活性有关,但大多数有丝分裂磷酸酯的作用仍不清楚。在这项研究中,我们检查了有丝分裂的磷酸位点和邻近残基的结构特性,以了解重磷酸化在非结构域中的作用。对有丝分裂停滞和未停滞的HeLa细胞的定量质谱分析表明,在有丝分裂进入时,分别有> 4100和> 2200个残基被显着磷酸化或去磷酸化。计算的相邻单个磷酸位点的氨基酸序列的失调分数显示,> 70%的去磷酸化磷酸位点存在于无序区域,而50%的磷酸化位点存在于非结构域中。在非结构域中的磷酸化概率与有丝分裂中的磷酸化增量之间观察到明显的负相关。这些结果表明,在进入有丝分裂时,大量的磷酸基团从非结构化结构域中除去,并转移到结构化较大的结构域中。基因本体术语分析显示,与有丝分裂相关的蛋白质被严重磷酸化,而与RNA相关的蛋白质既被去磷酸化又被磷酸化,
更新日期:2019-10-30
中文翻译:
定量蛋白质组学表明有丝分裂的磷酸化/去磷酸化与底物的非结构化区域有很强的相关性。
蛋白质磷酸化在有丝分裂的调控和进程中起着至关重要的作用。迄今为止,通过蛋白质组学分析已鉴定出> 40,000个磷酸化残基和相关的激酶。尽管这些磷酸酯中的一些与调节蛋白质-蛋白质相互作用或底物蛋白的催化活性有关,但大多数有丝分裂磷酸酯的作用仍不清楚。在这项研究中,我们检查了有丝分裂的磷酸位点和邻近残基的结构特性,以了解重磷酸化在非结构域中的作用。对有丝分裂停滞和未停滞的HeLa细胞的定量质谱分析表明,在有丝分裂进入时,分别有> 4100和> 2200个残基被显着磷酸化或去磷酸化。计算的相邻单个磷酸位点的氨基酸序列的失调分数显示,> 70%的去磷酸化磷酸位点存在于无序区域,而50%的磷酸化位点存在于非结构域中。在非结构域中的磷酸化概率与有丝分裂中的磷酸化增量之间观察到明显的负相关。这些结果表明,在进入有丝分裂时,大量的磷酸基团从非结构化结构域中除去,并转移到结构化较大的结构域中。基因本体术语分析显示,与有丝分裂相关的蛋白质被严重磷酸化,而与RNA相关的蛋白质既被去磷酸化又被磷酸化,


























 京公网安备 11010802027423号
京公网安备 11010802027423号