Computational and Theoretical Chemistry ( IF 2.8 ) Pub Date : 2019-09-24 , DOI: 10.1016/j.comptc.2019.112588 Gaurav Srivastav , Bhoopendra Yadav , Rohit Kumar Yadav , R.A. Yadav
|
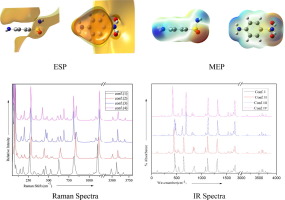
Molecular structures and vibrational parameters for all the four possible conformers of sulfanilamide were studied using Gaussian 09 software and PEDs were calculated using GAR2PED software. MEP and HOMO-LUMO energies were computed and NBO analysis was carried out. Optimized geometries possess Cs symmetry. Out of 51 normal modes, 26 modes are conformer sensitive, out of which 2 modes show frequency variation above 75 cm−1 in going from one conformer to another. Out of the 6 internal modes of NH2, 4 modes were found at lower frequencies for S-NH2 group compared to C-NH2 group, due to presence of intra-molecular O⋯H bonds in the SO2(NH2) group. Strength of nucleophilic attack is stronger with the H atoms of C-NH2 group compared to the H atoms of the S-NH2 group. Intra-molecular O⋯H bonds also lead to difference in corresponding geometrical parameters of S-NH2 and C-NH2 groups and difference in atomic charges at corresponding sites.
中文翻译:
DFT研究磺胺的分子结构构象和振动特性
使用高斯09软件研究了磺胺的所有四个可能构象异构体的分子结构和振动参数,并使用GAR2PED软件计算了PED。计算MEP和HOMO-LUMO能量,并进行NBO分析。优化的几何形状具有C s对称性。在51种正常模式中,有26种模式对众筹敏感,其中2种模式在从一个众筹者过渡到另一种众筹时表现出高于75 cm -1的频率变化。出NH的6种内部模式2,在对S-NH较低的频率中发现的4种模式2组相比C-NH 2基团,由于在SO分子内ö⋯H键存在2(NH 2) 团体。与S-NH 2基团的H原子相比,C-NH 2基团的H原子的亲核攻击强度更强。分子内的O⋯H键还导致S-NH 2和C-NH 2基团的相应几何参数的差异以及相应位点的原子电荷的差异。


























 京公网安备 11010802027423号
京公网安备 11010802027423号