当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Single‐reference coupled cluster methods for computing excitation energies in large molecules: The efficiency and accuracy of approximations
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 11.4 ) Pub Date : 2019-09-12 , DOI: 10.1002/wcms.1445 Róbert Izsák 1
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 11.4 ) Pub Date : 2019-09-12 , DOI: 10.1002/wcms.1445 Róbert Izsák 1
Affiliation
|
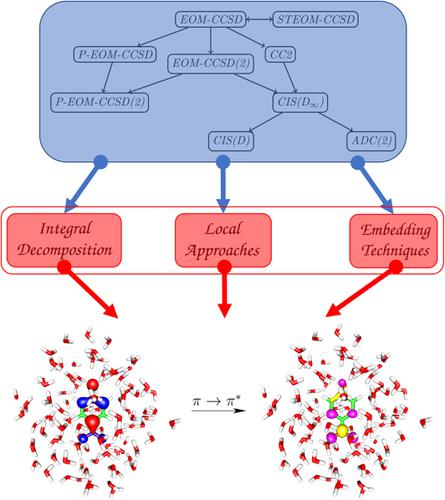
While methodological developments in the last decade made it possible to compute coupled cluster (CC) energies including excitations up to a perturbative triples correction for molecules containing several hundred atoms, a similar breakthrough has not yet been reported for excited state computations. Accurate CC methods for excited states are still expensive, although some promising candidates for an efficient and accurate excited state CC method have emerged recently. This review examines the various approximation schemes with particular emphasis on their performance for excitation energies and summarizes the best state‐of‐the‐art results which may pave the way for a robust excited state method applicable to molecules of hundreds of atoms. Among these, special attention will be given to exploiting the techniques of similarity transformation, perturbative approximations as well as integral decomposition, local and embedding techniques within the equation of motion CC framework.
中文翻译:

用于计算大分子激发能的单参考耦合簇方法:近似的效率和准确性
尽管近十年来方法学的发展使得可以计算耦合簇(CC)能量,包括对包含数百个原子的分子进行扰动三重校正的激发,但尚未为激发态计算报道类似的突破。尽管最近出现了一些有效而准确的激发态CC方法的有希望的候选方法,但用于激发态的精确CC方法仍然很昂贵。这篇综述对各种近似方案进行了研究,特别强调了它们对激发能的性能,并总结了最佳的最新技术成果,这可能为适用于数百个原子分子的鲁棒激发态方法铺平了道路。其中,将特别注意利用相似度转换技术,
更新日期:2019-09-12
中文翻译:
用于计算大分子激发能的单参考耦合簇方法:近似的效率和准确性
尽管近十年来方法学的发展使得可以计算耦合簇(CC)能量,包括对包含数百个原子的分子进行扰动三重校正的激发,但尚未为激发态计算报道类似的突破。尽管最近出现了一些有效而准确的激发态CC方法的有希望的候选方法,但用于激发态的精确CC方法仍然很昂贵。这篇综述对各种近似方案进行了研究,特别强调了它们对激发能的性能,并总结了最佳的最新技术成果,这可能为适用于数百个原子分子的鲁棒激发态方法铺平了道路。其中,将特别注意利用相似度转换技术,

























 京公网安备 11010802027423号
京公网安备 11010802027423号