Cell Death Discovery ( IF 7 ) Pub Date : 2019-07-19 , DOI: 10.1038/s41420-019-0198-y Qi Wang 1 , Song Wei 1, 2 , Haoming Zhou 1 , Gefenqiang Shen 1 , Xiaojie Gan 1 , Shun Zhou 1 , Jiannan Qiu 1 , Chenyu Shi 1 , Ling Lu 1, 2
|
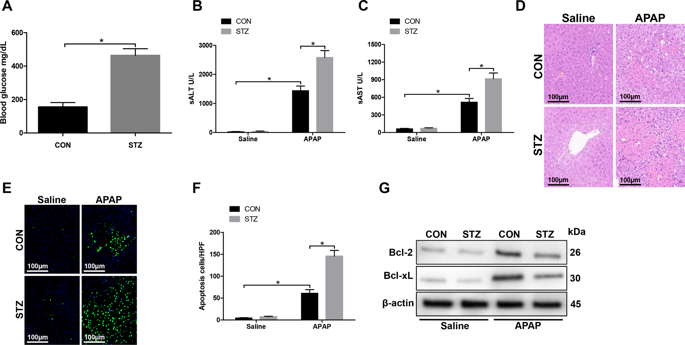
Although diabetes mellitus/hyperglycemia is a risk factor for acute liver injury, the underlying mechanism remains largely unknown. Liver-resident macrophages (Kupffer cells, KCs) and oxidative stress play critical roles in the pathogenesis of toxin-induced liver injury. Here, we evaluated the role of oxidative stress in regulating KC polarization against acetaminophen (APAP)-mediated acute liver injury in a streptozotocin-induced hyperglycemic murine model. Compared to the controls, hyperglycemic mice exhibited a significant increase in liver injury and intrahepatic inflammation. KCs obtained from hyperglycemic mice secreted higher levels of the proinflammatory factors, such as TNF-α and IL-6, lower levels of the anti-inflammatory factor IL-10. Furthermore, enhanced oxidative stress was revealed by increased levels of reactive oxygen species (ROS) in KCs from hyperglycemic mice post APAP treatment. In addition, ROS inhibitor NAC resulted in a significant decrease of ROS production in hyperglycemic KCs from mice posttreated with APAP. We also analyzed the role of hyperglycemia in macrophage M1/M2 polarization. Interestingly, we found that hyperglycemia promoted M1 polarization, but inhibited M2 polarization of KCs obtained from APAP-exposed livers, as evidenced by increased MCP-1 and inducible NO synthase (iNOS) gene induction but decreased Arg-1 and CD206 gene induction accompanied by increased STAT1 activation and decreased STAT6 activation. NAC restored Arg-1, CD206 gene induction, and STAT6 activation. To explore the mechanism how hyperglycemia regulates KCs polarization against APAP-induced acute liver injury, we examined the AMPK/PI3K/AKT signaling pathway and found decreased AMPK activation and increased AKT activation in liver and KCs from hyperglycemic mice post APAP treatment. AMPK activation by its agonist AICAR or PI3K inhibition by its antagonist LY294002 inhibited ROS production in KCs from hyperglycemic mice post APAP treatment and significantly attenuated APAP-induced liver injury in the hyperglycemic mice, compared to the control mice. Our results demonstrated that hyperglycemia exacerbated APAP-induced acute liver injury by promoting liver-resident macrophage proinflammatory response via AMPK/PI3K/AKT-mediated oxidative stress.
中文翻译:
高血糖症通过促进肝脏驻留巨噬细胞通过AMPK / PI3K / AKT介导的氧化应激的促炎反应来加剧对乙酰氨基酚引起的急性肝损伤
尽管糖尿病/高血糖是急性肝损伤的危险因素,但其潜在机制仍是未知之数。肝脏常驻巨噬细胞(枯否细胞,KCs)和氧化应激在毒素诱导的肝损伤的发病机理中起着至关重要的作用。在这里,我们评估了在链脲佐菌素诱导的高血糖小鼠模型中,氧化应激在调节KC极化对乙酰氨基酚(APAP)介导的急性肝损伤中的作用。与对照组相比,高血糖小鼠肝损伤和肝内炎症显着增加。从高血糖小鼠获得的KC分泌较高水平的促炎因子,例如TNF-α和IL-6,较低水平的抗炎因子IL-10。此外,在APAP治疗后,高血糖小鼠KC中的活性氧(ROS)水平升高,揭示了氧化应激增强。另外,ROS抑制剂NAC导致用APAP后处理的小鼠的高血糖KC中ROS产生的显着降低。我们还分析了高血糖在巨噬细胞M1 / M2极化中的作用。有趣的是,我们发现高血糖促进了从暴露于APAP的肝脏中获得的KCs的M1极化,但抑制了M2极化,这可通过MCP-1和诱导型一氧化氮合酶(iNOS)基因诱导增加,但Arg-1和CD206基因诱导伴随减少而增加STAT1激活增加而STAT6激活减少。NAC恢复了Arg-1,CD206基因的诱导和STAT6的激活。为了探索高血糖如何调节针对APAP诱导的急性肝损伤的KCs极化的机制,我们检查了AMPK / PI3K / AKT信号通路,发现高血糖小鼠APAP治疗后肝脏和KCs的AMPK激活降低和AKT激活增加。与对照小鼠相比,通过激动剂AICAR激活的AMPK或通过其拮抗剂LY294002抑制的PI3K抑制了高血糖小鼠在APAP治疗后KC中的ROS生成,并显着减轻了高血糖小鼠中APAP诱导的肝损伤。我们的结果表明,高血糖症通过通过AMPK / PI3K / AKT介导的氧化应激促进肝驻留巨噬细胞的促炎反应,加剧了APAP诱导的急性肝损伤。我们检查了AMPK / PI3K / AKT信号传导途径,发现APAP治疗后高血糖小鼠肝脏和KCs中AMPK激活减少,而AKT激活增加。与对照小鼠相比,通过激动剂AICAR激活的AMPK或通过其拮抗剂LY294002抑制的PI3K抑制了高血糖小鼠在APAP治疗后KC中的ROS生成,并显着减轻了高血糖小鼠中APAP诱导的肝损伤。我们的结果表明,高血糖症通过通过AMPK / PI3K / AKT介导的氧化应激促进肝驻留巨噬细胞的促炎反应,加剧了APAP诱导的急性肝损伤。我们检查了AMPK / PI3K / AKT信号传导途径,发现APAP治疗后高血糖小鼠肝脏和KCs中AMPK激活减少,而AKT激活增加。与对照小鼠相比,通过激动剂AICAR激活的AMPK或通过其拮抗剂LY294002抑制的PI3K抑制了高血糖小鼠在APAP治疗后KC中的ROS生成,并显着减轻了高血糖小鼠中APAP诱导的肝损伤。我们的结果表明,高血糖症通过通过AMPK / PI3K / AKT介导的氧化应激促进肝驻留巨噬细胞的促炎反应,加剧了APAP诱导的急性肝损伤。与对照小鼠相比,通过激动剂AICAR激活的AMPK或通过其拮抗剂LY294002抑制的PI3K抑制了高血糖小鼠在APAP治疗后KC中的ROS生成,并显着减轻了高血糖小鼠中APAP诱导的肝损伤。我们的结果表明,高血糖症通过通过AMPK / PI3K / AKT介导的氧化应激促进肝驻留巨噬细胞的促炎反应,加剧了APAP诱导的急性肝损伤。与对照小鼠相比,通过激动剂AICAR激活的AMPK或通过其拮抗剂LY294002抑制的PI3K抑制了高血糖小鼠在APAP治疗后KC中的ROS生成,并显着减轻了高血糖小鼠中APAP诱导的肝损伤。我们的结果表明,高血糖症通过通过AMPK / PI3K / AKT介导的氧化应激促进肝驻留巨噬细胞的促炎反应,加剧了APAP诱导的急性肝损伤。

























 京公网安备 11010802027423号
京公网安备 11010802027423号