当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
The electronic structure underlying electrocatalysis of two‐dimensional materials
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 11.4 ) Pub Date : 2019-05-07 , DOI: 10.1002/wcms.1418 Xunhua Zhao 1 , Jianjian Shi 1 , Yujin Ji 1 , Yuanyue Liu 1
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 11.4 ) Pub Date : 2019-05-07 , DOI: 10.1002/wcms.1418 Xunhua Zhao 1 , Jianjian Shi 1 , Yujin Ji 1 , Yuanyue Liu 1
Affiliation
|
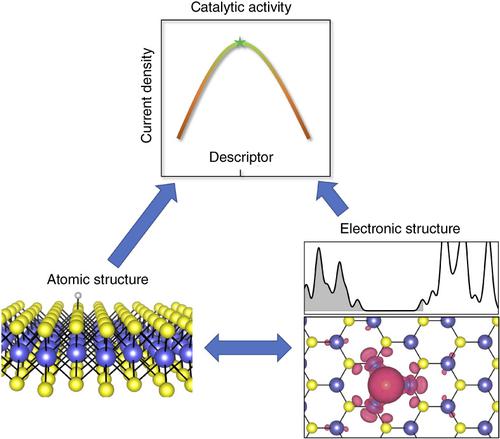
The understanding and development of advanced electrochemical catalysts have attracted intensive studies and achieved tremendous progress in the past decades. Two‐dimensional (2D) materials, such as doped graphene and atomically‐thin transition metal compounds, have shown great promise as electrocatalysts for various renewable energy conversion and storage reactions. Their further developments require improved understanding of the catalytic mechanisms at atomic level. Currently, most of the understandings are based on the formation free energies of the intermediates, which are determined by their binding strengths with the catalyst, usually calculated from density functional theory (DFT). These energies/binding strengths have been used as descriptor to describe the activity of many catalysts. However, it remains less explored why different catalysts have different binding strengths and what are the underlying factors controlling them, requiring studies going beyond atomic level to electronic level. This review aims to provide such links, focusing on 2D electrocatalysts for hydrogen evolution reaction (HER), oxygen reduction reaction/oxygen evolution reaction (ORR/OER), CO2 reduction reaction (CO2R) and nitrogen reduction reaction (NRR). We also discuss some of the significant issues that need to be addressed in DFT calculations, including the effects of varying charge and fixed potential of the catalyst, the passivation of active sites, and the solvation effects.
中文翻译:

二维材料电催化基础的电子结构
在过去的几十年中,对先进电化学催化剂的理解和发展吸引了广泛的研究并取得了巨大的进步。二维(2D)材料(例如掺杂的石墨烯和原子稀薄的过渡金属化合物)作为用于各种可再生能源转化和存储反应的电催化剂,显示出巨大的希望。它们的进一步发展需要在原子水平上更好地理解催化机理。当前,大多数理解是基于中间体的形成自由能,该自由能由它们与催化剂的结合强度决定,通常由密度泛函理论(DFT)计算得出。这些能量/结合强度已被用作描述许多催化剂活性的描述符。然而,为何不同的催化剂具有不同的结合强度以及控制它们的潜在因素还有待探讨,因此需要进行从原子级到电子级的研究。这篇综述旨在提供这样的联系,重点是用于氢析出反应(HER),氧还原反应/氧析出反应(ORR / OER),CO2还原反应(CO2R)和氮还原反应(NRR)的2D电催化剂。我们还讨论了DFT计算中需要解决的一些重要问题,包括催化剂电荷和固定电势的变化,活性部位的钝化和溶剂化效应的影响。这篇综述旨在提供这样的联系,重点是用于氢析出反应(HER),氧还原反应/氧析出反应(ORR / OER),CO2还原反应(CO2R)和氮还原反应(NRR)的2D电催化剂。我们还讨论了DFT计算中需要解决的一些重要问题,包括催化剂电荷和固定电势的变化,活性部位的钝化和溶剂化效应的影响。这篇综述旨在提供这样的联系,重点是用于氢析出反应(HER),氧还原反应/氧析出反应(ORR / OER),CO2还原反应(CO2R)和氮还原反应(NRR)的2D电催化剂。我们还讨论了DFT计算中需要解决的一些重要问题,包括催化剂电荷和固定电势的变化,活性部位的钝化和溶剂化效应的影响。
更新日期:2019-05-16
中文翻译:
二维材料电催化基础的电子结构
在过去的几十年中,对先进电化学催化剂的理解和发展吸引了广泛的研究并取得了巨大的进步。二维(2D)材料(例如掺杂的石墨烯和原子稀薄的过渡金属化合物)作为用于各种可再生能源转化和存储反应的电催化剂,显示出巨大的希望。它们的进一步发展需要在原子水平上更好地理解催化机理。当前,大多数理解是基于中间体的形成自由能,该自由能由它们与催化剂的结合强度决定,通常由密度泛函理论(DFT)计算得出。这些能量/结合强度已被用作描述许多催化剂活性的描述符。然而,为何不同的催化剂具有不同的结合强度以及控制它们的潜在因素还有待探讨,因此需要进行从原子级到电子级的研究。这篇综述旨在提供这样的联系,重点是用于氢析出反应(HER),氧还原反应/氧析出反应(ORR / OER),CO2还原反应(CO2R)和氮还原反应(NRR)的2D电催化剂。我们还讨论了DFT计算中需要解决的一些重要问题,包括催化剂电荷和固定电势的变化,活性部位的钝化和溶剂化效应的影响。这篇综述旨在提供这样的联系,重点是用于氢析出反应(HER),氧还原反应/氧析出反应(ORR / OER),CO2还原反应(CO2R)和氮还原反应(NRR)的2D电催化剂。我们还讨论了DFT计算中需要解决的一些重要问题,包括催化剂电荷和固定电势的变化,活性部位的钝化和溶剂化效应的影响。这篇综述旨在提供这样的联系,重点是用于氢析出反应(HER),氧还原反应/氧析出反应(ORR / OER),CO2还原反应(CO2R)和氮还原反应(NRR)的2D电催化剂。我们还讨论了DFT计算中需要解决的一些重要问题,包括催化剂电荷和固定电势的变化,活性部位的钝化和溶剂化效应的影响。


























 京公网安备 11010802027423号
京公网安备 11010802027423号