Structure ( IF 5.7 ) Pub Date : 2023-04-28 , DOI: 10.1016/j.str.2023.04.005 Hope Woods 1 , Dominic L Schiano 2 , Jonathan I Aguirre 3 , Kaitlyn V Ledwitch 2 , Eli F McDonald 2 , Markus Voehler 2 , Jens Meiler 4 , Clara T Schoeder 4
|
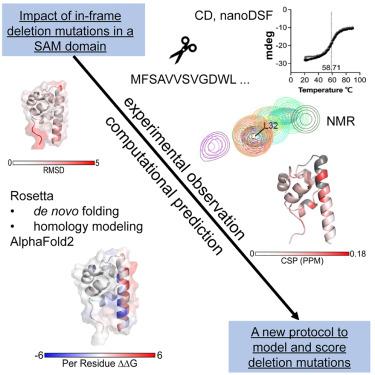
In-frame deletion mutations can result in disease. The impact of these mutations on protein structure and subsequent functional changes remain understudied, partially due to the lack of comprehensive datasets including a structural readout. In addition, the recent breakthrough in structure prediction through deep learning demands an update of computational deletion mutation prediction. In this study, we deleted individually every residue of a small α-helical sterile alpha motif domain and investigated the structural and thermodynamic changes using 2D NMR spectroscopy and differential scanning fluorimetry. Then, we tested computational protocols to model and classify observed deletion mutants. We show a method using AlphaFold2 followed by RosettaRelax performs the best overall. In addition, a metric containing pLDDT values and Rosetta ΔΔG is most reliable in classifying tolerated deletion mutations. We further test this method on other datasets and show they hold for proteins known to harbor disease-causing deletion mutations.
中文翻译:
缺失突变体的计算建模和预测
框内缺失突变可导致疾病。这些突变对蛋白质结构和随后的功能变化的影响仍未得到充分研究,部分原因是缺乏包括结构读数在内的综合数据集。此外,最近通过深度学习在结构预测方面取得的突破要求更新计算缺失突变预测。在这项研究中,我们分别删除了一个小的 α-螺旋无菌 α 基序结构域的每个残基,并使用 2D NMR 光谱和差示扫描荧光测定法研究了结构和热力学变化。然后,我们测试了计算协议来对观察到的缺失突变体进行建模和分类。我们展示了一种使用 AlphaFold2 后跟 RosettaRelax 的方法,其整体效果最好。此外,包含 pLDDT 值和 Rosetta ΔΔG 的指标在对可耐受的缺失突变进行分类时最可靠。我们在其他数据集上进一步测试了这种方法,并表明它们适用于已知含有致病缺失突变的蛋白质。
























 京公网安备 11010802027423号
京公网安备 11010802027423号