当前位置:
X-MOL 学术
›
Ind. Eng. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Interpretable Machine Learning Model for Predicting Interaction Energies between Dimethyl Sulfide and Potential Absorbing Solvents
Industrial & Engineering Chemistry Research ( IF 4.2 ) Pub Date : 2023-03-13 , DOI: 10.1021/acs.iecr.2c04559 Chuanlei Liu, Yuxiang Chen, Guanchu Guo, Qiyue Zhao, Hao Jiang, Kongguo Wu, Qilong Peng, Yu Chen, Diyi Fang, Benxian Shen, Haitao Shen, Di Wu, Hui Sun
Industrial & Engineering Chemistry Research ( IF 4.2 ) Pub Date : 2023-03-13 , DOI: 10.1021/acs.iecr.2c04559 Chuanlei Liu, Yuxiang Chen, Guanchu Guo, Qiyue Zhao, Hao Jiang, Kongguo Wu, Qilong Peng, Yu Chen, Diyi Fang, Benxian Shen, Haitao Shen, Di Wu, Hui Sun
|
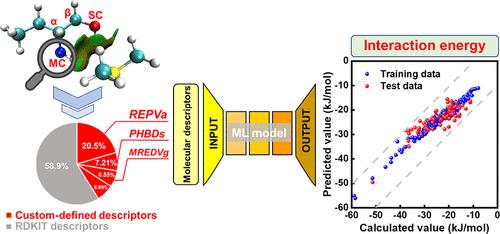
Non-bonding intermolecular interactions largely dominate the selective dissolution of trace species into physical solvents and, therefore, are fundamentally important to solvent development for the capture of environment-undesired compounds or purification of chemicals. However, acquirement of the interaction energy requires costly quantum chemical computation and still encounters a practical challenge to build a chemically interpretable machine learning (ML) prediction model using documented molecular descriptors. Herein, we report an ML model for predicting the interaction energies (Eint) between dimethyl sulfide and potential absorbing solvents. Applying the reduced density gradient and quantum theory of atoms in molecules analyses, the non-bonding intermolecular interactions of dimethyl sulfide with solvent compounds were elucidated through focusing on the molecular fragments containing the main center (MC) and secondary center (SC) rather than the whole molecule. The training data set was obtained using a molecular generation strategy, and 21 molecular descriptors were defined to describe the electronic states of the central atoms in each solvent molecule and its nearby hydrogen-bond donors. Model analysis reveals that Eint is mainly determined by the charge state of the crucial fragments and the hydrogen-bond donors of the solvent molecule. The custom-defined descriptors not only improve the regression and prediction performance but also enable the interpretability of the ML model. Additionally, the absorption equilibrium measurements of solubilities of dimethyl sulfide in several commercial solvents verified the strong correlation between the dissolving affinity and solute–solvent intermolecular interaction energy. The present study provides an approach to building practical and interpretable intelligent algorithms to aid the development of sustainable chemicals or processes.
中文翻译:

用于预测二甲基硫醚与潜在吸收溶剂之间相互作用能的可解释机器学习模型
非键合分子间相互作用在很大程度上决定了痕量物质选择性溶解到物理溶剂中,因此,对于用于捕获环境不需要的化合物或纯化化学品的溶剂开发至关重要。然而,相互作用能的获取需要昂贵的量子化学计算,并且在使用记录的分子描述符构建化学可解释的机器学习 (ML) 预测模型方面仍然面临实际挑战。在此,我们报告了一个用于预测相互作用能量的 ML 模型 ( E int) 在二甲硫醚和潜在的吸收溶剂之间。应用分子分析中的约化密度梯度和原子量子理论,通过关注包含主中心(MC)和次中心(SC)的分子片段而不是整个分子。训练数据集是使用分子生成策略获得的,并定义了 21 个分子描述符来描述每个溶剂分子及其附近氢键供体中中心原子的电子态。模型分析表明,E int主要由关键片段的电荷状态和溶剂分子的氢键供体决定。自定义描述符不仅可以提高回归和预测性能,还可以实现 ML 模型的可解释性。此外,二甲硫醚在几种商业溶剂中溶解度的吸收平衡测量证实了溶解亲和力与溶质-溶剂分子间相互作用能之间的强相关性。本研究提供了一种构建实用且可解释的智能算法的方法,以帮助开发可持续的化学品或工艺。
更新日期:2023-03-13
中文翻译:
用于预测二甲基硫醚与潜在吸收溶剂之间相互作用能的可解释机器学习模型
非键合分子间相互作用在很大程度上决定了痕量物质选择性溶解到物理溶剂中,因此,对于用于捕获环境不需要的化合物或纯化化学品的溶剂开发至关重要。然而,相互作用能的获取需要昂贵的量子化学计算,并且在使用记录的分子描述符构建化学可解释的机器学习 (ML) 预测模型方面仍然面临实际挑战。在此,我们报告了一个用于预测相互作用能量的 ML 模型 ( E int) 在二甲硫醚和潜在的吸收溶剂之间。应用分子分析中的约化密度梯度和原子量子理论,通过关注包含主中心(MC)和次中心(SC)的分子片段而不是整个分子。训练数据集是使用分子生成策略获得的,并定义了 21 个分子描述符来描述每个溶剂分子及其附近氢键供体中中心原子的电子态。模型分析表明,E int主要由关键片段的电荷状态和溶剂分子的氢键供体决定。自定义描述符不仅可以提高回归和预测性能,还可以实现 ML 模型的可解释性。此外,二甲硫醚在几种商业溶剂中溶解度的吸收平衡测量证实了溶解亲和力与溶质-溶剂分子间相互作用能之间的强相关性。本研究提供了一种构建实用且可解释的智能算法的方法,以帮助开发可持续的化学品或工艺。
























 京公网安备 11010802027423号
京公网安备 11010802027423号