当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Structural Phase Transition of Quinone-Containing PAH Derivatives on Au(111) at High Coverage
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2023-03-03 , DOI: 10.1021/acs.jpcc.2c08811 Xin Song 1 , Zhicun Zhou 2 , Xinchen Peng 2 , Xinbang Liu 2 , Dianrong Han 1 , Hang Zhang 2 , Xiaoqing Liu 3 , Li Wang 3 , Huihui Kong 2
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2023-03-03 , DOI: 10.1021/acs.jpcc.2c08811 Xin Song 1 , Zhicun Zhou 2 , Xinchen Peng 2 , Xinbang Liu 2 , Dianrong Han 1 , Hang Zhang 2 , Xiaoqing Liu 3 , Li Wang 3 , Huihui Kong 2
Affiliation
|
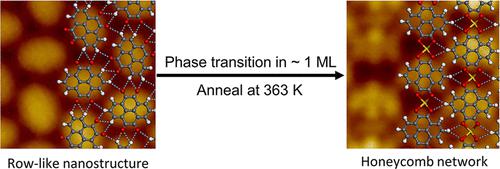
Delicate control over structural phase transition provides advanced approaches for the fabrication of the desired well-ordered nanoarchitectures on surfaces. The participation of intrinsic metal adatoms in pure organic systems can facilitate the structural phase transition by direct capture of surface metal adatoms and forming metal–organic bonds. However, most of the situations occur at low coverage; such structural phase transition at a higher molecular concentration is limited to some extent due to the poor migration ability. Thus, high-concentration phase transition needs to be explored, which might be significant for the design and exploitation of large-scale ordered metal–organic-related nanomaterials. Herein, we report the phase transition of pyrene-4,5,9,10-tetraone (PT) molecules at high coverage (∼1 monolayer (ML)) on Au(111) from hydrogen-bonded row-like nanostructures to metal–organic honeycomb networks by coordinating with surface Au adatoms as demonstrated by scanning tunneling microscopy (STM). Combined with density functional theory (DFT) calculations, we demonstrate that identical molecular density (or unit cells) of two nanostructures should be the key, which makes it possible to realize phase transition possibly by in situ rotation and coordinating with the gold adatoms. Also, the phase transition causes the modulation of electronic properties from semiconductive ones to metallic ones.
中文翻译:

含醌多环芳烃衍生物在 Au(111) 上高覆盖率的结构相变
对结构相变的精细控制为在表面上制造所需的有序纳米结构提供了先进的方法。纯有机体系中固有金属吸附原子的参与可以通过直接捕获表面金属吸附原子并形成金属-有机键来促进结构相变。但是,大多数情况都发生在低覆盖率的情况下;由于迁移能力差,这种在较高分子浓度下的结构相变在一定程度上受到限制。因此,需要探索高浓度相变,这对于设计和开发大规模有序金属-有机相关纳米材料可能具有重要意义。在这里,我们报告了芘-4,5,9 的相变,10-四酮 (PT) 分子在 Au(111) 上的高覆盖度(~1 单层 (ML))从氢键键合的行状纳米结构到金属有机蜂窝网络,通过与表面 Au 吸附原子协调,如扫描隧道显微镜所示( ST)。结合密度泛函理论 (DFT) 计算,我们证明了两个纳米结构的相同分子密度(或晶胞)应该是关键,这使得通过原位旋转和与金吸附原子配位实现相变成为可能。此外,相变导致电子特性从半导体特性到金属特性的调制。结合密度泛函理论 (DFT) 计算,我们证明了两个纳米结构的相同分子密度(或晶胞)应该是关键,这使得通过原位旋转和与金吸附原子配位实现相变成为可能。此外,相变导致电子特性从半导体特性到金属特性的调制。结合密度泛函理论 (DFT) 计算,我们证明了两个纳米结构的相同分子密度(或晶胞)应该是关键,这使得通过原位旋转和与金吸附原子配位实现相变成为可能。此外,相变导致电子特性从半导体特性到金属特性的调制。
更新日期:2023-03-03
中文翻译:
含醌多环芳烃衍生物在 Au(111) 上高覆盖率的结构相变
对结构相变的精细控制为在表面上制造所需的有序纳米结构提供了先进的方法。纯有机体系中固有金属吸附原子的参与可以通过直接捕获表面金属吸附原子并形成金属-有机键来促进结构相变。但是,大多数情况都发生在低覆盖率的情况下;由于迁移能力差,这种在较高分子浓度下的结构相变在一定程度上受到限制。因此,需要探索高浓度相变,这对于设计和开发大规模有序金属-有机相关纳米材料可能具有重要意义。在这里,我们报告了芘-4,5,9 的相变,10-四酮 (PT) 分子在 Au(111) 上的高覆盖度(~1 单层 (ML))从氢键键合的行状纳米结构到金属有机蜂窝网络,通过与表面 Au 吸附原子协调,如扫描隧道显微镜所示( ST)。结合密度泛函理论 (DFT) 计算,我们证明了两个纳米结构的相同分子密度(或晶胞)应该是关键,这使得通过原位旋转和与金吸附原子配位实现相变成为可能。此外,相变导致电子特性从半导体特性到金属特性的调制。结合密度泛函理论 (DFT) 计算,我们证明了两个纳米结构的相同分子密度(或晶胞)应该是关键,这使得通过原位旋转和与金吸附原子配位实现相变成为可能。此外,相变导致电子特性从半导体特性到金属特性的调制。结合密度泛函理论 (DFT) 计算,我们证明了两个纳米结构的相同分子密度(或晶胞)应该是关键,这使得通过原位旋转和与金吸附原子配位实现相变成为可能。此外,相变导致电子特性从半导体特性到金属特性的调制。


























 京公网安备 11010802027423号
京公网安备 11010802027423号