当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Demystifying the Chemical Ordering of Multimetallic Nanoparticles
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2023-01-21 , DOI: 10.1021/acs.accounts.2c00646 Dennis Johan Loevlie 1 , Brenno Ferreira 1 , Giannis Mpourmpakis 1
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2023-01-21 , DOI: 10.1021/acs.accounts.2c00646 Dennis Johan Loevlie 1 , Brenno Ferreira 1 , Giannis Mpourmpakis 1
Affiliation
|
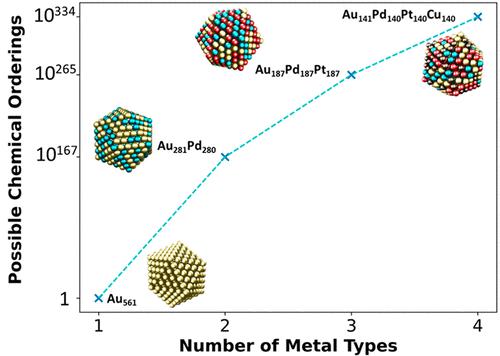
Multimetallic nanoparticles (NPs) have highly tunable properties due to the synergy between the different metals and the wide variety of NP structural parameters such as size, shape, composition, and chemical ordering. The major problem with studying multimetallic NPs is that as the number of different metals increases, the number of possible chemical orderings (placements of different metals) for a NP of fixed size explodes. Thus, it becomes infeasible to explore NP energetic differences with highly accurate computational methods, such as density functional theory (DFT), which has a high computational cost and is typically applied to up to a couple of hundred metal atoms. Here, we demonstrate a methodology advancing NP simulations by effectively exploring the vast materials space of multimetallic NPs and accurately identifying the ones with the most thermodynamically preferred chemical orderings. With accuracies reaching that of DFT, our methodology is applicable to practically any NP size, shape, and metal composition. We achieve this by significantly advancing the bond-centric (BC) model, a physics-based model that has been previously shown to rapidly predict bimetallic NP cohesive energies (CEs). Specifically, the BC model is trained in a way to understand how the bimetallic bond strength changes under different coordination environments present on a NP and how the metal composition of every site affects the detailed coordination environment using fractional coordination numbers. This newly modified BC model leads to an improvement from 0.331 (original model) to 0.089 eV/atom in CE predictions when compared to DFT values on a robust data set of 90 different NPs consisting of PtPd, AuPt, and AuPd NPs with varying compositions and chemical orderings. By incorporating the modified BC model into an in-house-developed genetic algorithm (GA) we can effectively and accurately predict the most stable chemical orderings of large, realistic bimetallic NPs consisting of thousands of metal atoms. This is demonstrated on AuPd bimetallic NPs, a challenging system due to the similarity in the cohesion of the two metals. By training our BC model using a unique DFT calculation on a bimetallic NP (one calculation for two metals combining together), we expand to explore the chemical ordering of multimetallic NPs. We first demonstrate the application of our methodology on a AuPdPt NP and validate our stability predictions with literature data. Then, we effectively explore the vast materials space of multimetallic NPs consisting of combinations of Au, Pt, and Pd as a function of metal composition. Our thermodynamic stability trends are presented in a ternary diagram revealing detailed, and yet, unexpected chemical ordering trends. Our computational framework can aid both experimental and computational researchers toward effectively screening multimetallic NP stability. Moreover, we provide an outlook of how this framework can be applied to catalyst discovery, high-entropy alloys, and single-atom alloys.
中文翻译:

揭秘多金属纳米粒子的化学排序
由于不同金属之间的协同作用以及各种 NP 结构参数(例如尺寸、形状、组成和化学排序),多金属纳米颗粒 (NP) 具有高度可调的特性。研究多金属 NP 的主要问题是,随着不同金属数量的增加,固定尺寸 NP 的可能化学排序(不同金属的排列)数量会激增。因此,用高精度计算方法探索 NP 能量差异变得不可行,例如密度泛函理论 (DFT),它具有很高的计算成本并且通常应用于多达几百个金属原子。这里,我们展示了一种通过有效探索多金属 NP 的广阔材料空间并准确识别具有最热力学首选化学顺序的材料来推进 NP 模拟的方法。由于精度达到 DFT,我们的方法适用于几乎任何 NP 尺寸、形状和金属成分。我们通过显着推进以键为中心 (BC) 模型来实现这一目标,该模型是一种基于物理学的模型,之前已被证明可以快速预测双金属 NP 内聚能 (CE)。具体来说,BC 模型以某种方式进行训练,以了解双金属键强度在 NP 上存在的不同配位环境下如何变化,以及每个位点的金属成分如何使用分数配位数影响详细的配位环境。与由 90 种不同 NP 组成的稳健数据集的 DFT 值相比,这种新修改的 BC 模型导致 CE 预测从 0.331(原始模型)提高到 0.089 eV/原子化学品订单。通过将修改后的 BC 模型整合到内部开发的遗传算法 (GA) 中,我们可以有效准确地预测由数千个金属原子组成的大型、逼真的双金属 NP 的最稳定化学排序。这在 AuPd 双金属 NPs 上得到了证明,这是一个具有挑战性的系统,因为两种金属的内聚力相似。通过在双金属 NP 上使用独特的 DFT 计算来训练我们的 BC 模型(两种金属组合在一起的一种计算),我们扩展以探索多金属 NP 的化学排序。我们首先展示了我们的方法在 AuPdPt NP 上的应用,并使用文献数据验证了我们的稳定性预测。然后,我们有效地探索了由 Au、Pt 和 Pd 的组合组成的多金属 NP 的广阔材料空间,作为金属成分的函数。我们的热力学稳定性趋势以三元图形式呈现,揭示了详细的、意想不到的化学排序趋势。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们有效地探索了由 Au、Pt 和 Pd 的组合组成的多金属 NP 的广阔材料空间,作为金属成分的函数。我们的热力学稳定性趋势以三元图形式呈现,揭示了详细的、意想不到的化学排序趋势。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们有效地探索了由 Au、Pt 和 Pd 的组合组成的多金属 NP 的广阔材料空间,作为金属成分的函数。我们的热力学稳定性趋势以三元图形式呈现,揭示了详细的、意想不到的化学排序趋势。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。
更新日期:2023-01-21
中文翻译:
揭秘多金属纳米粒子的化学排序
由于不同金属之间的协同作用以及各种 NP 结构参数(例如尺寸、形状、组成和化学排序),多金属纳米颗粒 (NP) 具有高度可调的特性。研究多金属 NP 的主要问题是,随着不同金属数量的增加,固定尺寸 NP 的可能化学排序(不同金属的排列)数量会激增。因此,用高精度计算方法探索 NP 能量差异变得不可行,例如密度泛函理论 (DFT),它具有很高的计算成本并且通常应用于多达几百个金属原子。这里,我们展示了一种通过有效探索多金属 NP 的广阔材料空间并准确识别具有最热力学首选化学顺序的材料来推进 NP 模拟的方法。由于精度达到 DFT,我们的方法适用于几乎任何 NP 尺寸、形状和金属成分。我们通过显着推进以键为中心 (BC) 模型来实现这一目标,该模型是一种基于物理学的模型,之前已被证明可以快速预测双金属 NP 内聚能 (CE)。具体来说,BC 模型以某种方式进行训练,以了解双金属键强度在 NP 上存在的不同配位环境下如何变化,以及每个位点的金属成分如何使用分数配位数影响详细的配位环境。与由 90 种不同 NP 组成的稳健数据集的 DFT 值相比,这种新修改的 BC 模型导致 CE 预测从 0.331(原始模型)提高到 0.089 eV/原子化学品订单。通过将修改后的 BC 模型整合到内部开发的遗传算法 (GA) 中,我们可以有效准确地预测由数千个金属原子组成的大型、逼真的双金属 NP 的最稳定化学排序。这在 AuPd 双金属 NPs 上得到了证明,这是一个具有挑战性的系统,因为两种金属的内聚力相似。通过在双金属 NP 上使用独特的 DFT 计算来训练我们的 BC 模型(两种金属组合在一起的一种计算),我们扩展以探索多金属 NP 的化学排序。我们首先展示了我们的方法在 AuPdPt NP 上的应用,并使用文献数据验证了我们的稳定性预测。然后,我们有效地探索了由 Au、Pt 和 Pd 的组合组成的多金属 NP 的广阔材料空间,作为金属成分的函数。我们的热力学稳定性趋势以三元图形式呈现,揭示了详细的、意想不到的化学排序趋势。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们有效地探索了由 Au、Pt 和 Pd 的组合组成的多金属 NP 的广阔材料空间,作为金属成分的函数。我们的热力学稳定性趋势以三元图形式呈现,揭示了详细的、意想不到的化学排序趋势。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们有效地探索了由 Au、Pt 和 Pd 的组合组成的多金属 NP 的广阔材料空间,作为金属成分的函数。我们的热力学稳定性趋势以三元图形式呈现,揭示了详细的、意想不到的化学排序趋势。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。我们的计算框架可以帮助实验和计算研究人员有效筛选多金属 NP 稳定性。此外,我们展望了该框架如何应用于催化剂发现、高熵合金和单原子合金。
























 京公网安备 11010802027423号
京公网安备 11010802027423号