当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Electronic-Structure Properties from Atom-Centered Predictions of the Electron Density
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-12-01 , DOI: 10.1021/acs.jctc.2c00850 Andrea Grisafi 1 , Alan M Lewis 2 , Mariana Rossi 2 , Michele Ceriotti 3
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-12-01 , DOI: 10.1021/acs.jctc.2c00850 Andrea Grisafi 1 , Alan M Lewis 2 , Mariana Rossi 2 , Michele Ceriotti 3
Affiliation
|
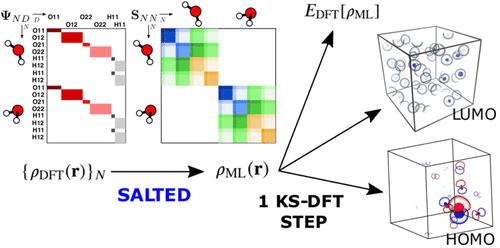
The electron density of a molecule or material has recently received major attention as a target quantity of machine-learning models. A natural choice to construct a model that yields transferable and linear-scaling predictions is to represent the scalar field using a multicentered atomic basis analogous to that routinely used in density fitting approximations. However, the nonorthogonality of the basis poses challenges for the learning exercise, as it requires accounting for all the atomic density components at once. We devise a gradient-based approach to directly minimize the loss function of the regression problem in an optimized and highly sparse feature space. In so doing, we overcome the limitations associated with adopting an atom-centered model to learn the electron density over arbitrarily complex data sets, obtaining very accurate predictions using a comparatively small training set. The enhanced framework is tested on 32-molecule periodic cells of liquid water, presenting enough complexity to require an optimal balance between accuracy and computational efficiency. We show that starting from the predicted density a single Kohn–Sham diagonalization step can be performed to access total energy components that carry an error of just 0.1 meV/atom with respect to the reference density functional calculations. Finally, we test our method on the highly heterogeneous QM9 benchmark data set, showing that a small fraction of the training data is enough to derive ground-state total energies within chemical accuracy.
中文翻译:

来自以原子为中心的电子密度预测的电子结构特性
分子或材料的电子密度最近作为机器学习模型的目标量受到了广泛关注。构建产生可转移和线性缩放预测的模型的自然选择是使用类似于密度拟合近似中常规使用的多中心原子基础来表示标量场。然而,基础的非正交性给学习练习带来了挑战,因为它需要同时考虑所有原子密度分量。我们设计了一种基于梯度的方法来直接最小化优化且高度稀疏的特征空间中回归问题的损失函数。这样做,我们克服了采用以原子为中心的模型来学习任意复杂数据集上的电子密度的局限性,使用相对较小的训练集获得了非常准确的预测。增强的框架在液态水的 32 分子周期细胞上进行了测试,表现出足够的复杂性,需要准确性和计算效率之间的最佳平衡。我们表明,从预测密度开始,可以执行单个 Kohn-Sham 对角化步骤来获取总能量分量,相对于参考密度泛函计算,其误差仅为 0.1 meV/原子。最后,我们在高度异构的 QM9 基准数据集上测试我们的方法,表明训练数据的一小部分足以在化学精度内推导出基态总能量。
更新日期:2022-12-01
中文翻译:
来自以原子为中心的电子密度预测的电子结构特性
分子或材料的电子密度最近作为机器学习模型的目标量受到了广泛关注。构建产生可转移和线性缩放预测的模型的自然选择是使用类似于密度拟合近似中常规使用的多中心原子基础来表示标量场。然而,基础的非正交性给学习练习带来了挑战,因为它需要同时考虑所有原子密度分量。我们设计了一种基于梯度的方法来直接最小化优化且高度稀疏的特征空间中回归问题的损失函数。这样做,我们克服了采用以原子为中心的模型来学习任意复杂数据集上的电子密度的局限性,使用相对较小的训练集获得了非常准确的预测。增强的框架在液态水的 32 分子周期细胞上进行了测试,表现出足够的复杂性,需要准确性和计算效率之间的最佳平衡。我们表明,从预测密度开始,可以执行单个 Kohn-Sham 对角化步骤来获取总能量分量,相对于参考密度泛函计算,其误差仅为 0.1 meV/原子。最后,我们在高度异构的 QM9 基准数据集上测试我们的方法,表明训练数据的一小部分足以在化学精度内推导出基态总能量。

























 京公网安备 11010802027423号
京公网安备 11010802027423号