当前位置:
X-MOL 学术
›
J. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Electronic and Steric Control of Rates and Selectivities in Rhodium-Catalyzed [2+2+2] Cycloadditions for Constructing Fused Tricyclic Hydronaphthofurans: A Density Functional Theory Study
The Journal of Organic Chemistry ( IF 3.6 ) Pub Date : 2022-11-30 , DOI: 10.1021/acs.joc.2c01937 Jiang-Ping Li 1 , Li-Juan Dou 1 , Wei-Hua Mu 1
The Journal of Organic Chemistry ( IF 3.6 ) Pub Date : 2022-11-30 , DOI: 10.1021/acs.joc.2c01937 Jiang-Ping Li 1 , Li-Juan Dou 1 , Wei-Hua Mu 1
Affiliation
|
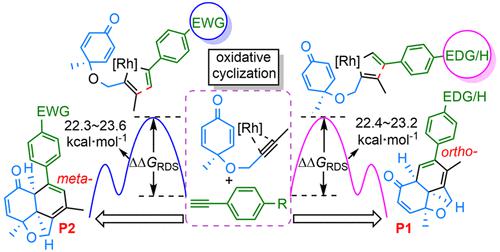
Fused tricyclic hydronaphthofurans with multiple chiral centers are very important skeletons for constructing natural products; however, their synthesis is challenging, and a detailed understanding of the final formation mechanism remains elusive. In this work, density functional theory computations were employed to characterize rhodium-catalyzed [2+2+2] cycloaddition of enyne with terminal alkynes. The putative mechanism involves an initial ligand exchange, followed by oxidative cyclization, olefin insertion, and reductive elimination processes. Oxidative cyclization is shown to be the rate- and selectivity-determining step of the full chemical transformation, where the R substituent on terminal alkynes has a significant influence on the reaction selectivities. When R is an electron-donating group (OMe and Me), the ortho-substituted tricyclic hydronaphthofurans (P1) are predicted to be dominant; on the contrary, meta-substituted compounds P2 emerge as the main products when R is an electron-withdrawing group (NO2, CF3, and CN). Computational predictions for selectivity are in good agreement with experimental product ratios. Free energy barriers of the rate-determining step for P1 and P2 are ∼22.3–23.6 kcal mol–1, which align well with their experimental yields of ∼79–92% at 313 K after 0.5 h. The results also accurately reproduce experimentally observed regio-, chemo-, and enantioselectivities, with steric hindrance as well as electronic properties of the substrate and ligand markedly influencing the reaction rates and selectivities. The influence of computational methods is also explored and discussed in detail.
中文翻译:

用于构建稠合三环萘并呋喃的铑催化 [2+2+2] 环加成反应中速率和选择性的电子和空间控制:密度泛函理论研究
具有多个手性中心的稠合三环氢化萘并呋喃是构建天然产物的重要骨架;然而,它们的合成具有挑战性,对最终形成机制的详细了解仍然难以捉摸。在这项工作中,采用密度泛函理论计算来表征铑催化的烯炔与末端炔烃的 [2+2+2] 环加成反应。推定的机制涉及初始配体交换,然后是氧化环化、烯烃插入和还原消除过程。氧化环化被证明是完全化学转化的速率和选择性决定步骤,其中末端炔烃上的 R 取代基对反应选择性具有显着影响。当 R 是给电子基团(OMe 和 Me)时,邻位-取代的三环萘氢呋喃 ( P1 ) 预计将占主导地位;相反,当R为吸电子基团(NO 2、CF 3和CN)时,间位取代化合物P2成为主要产物。选择性的计算预测与实验产品比率非常一致。P1和P2的决速步骤的自由能垒为∼22.3–23.6 kcal mol –1,这与他们在 313 K 下 0.5 小时后的实验产率为 ~79-92% 非常吻合。结果还准确地再现了实验观察到的区域、化学和对映选择性,空间位阻以及底物和配体的电子特性显着影响反应速率和选择性。还详细探讨和讨论了计算方法的影响。
更新日期:2022-11-30
中文翻译:
用于构建稠合三环萘并呋喃的铑催化 [2+2+2] 环加成反应中速率和选择性的电子和空间控制:密度泛函理论研究
具有多个手性中心的稠合三环氢化萘并呋喃是构建天然产物的重要骨架;然而,它们的合成具有挑战性,对最终形成机制的详细了解仍然难以捉摸。在这项工作中,采用密度泛函理论计算来表征铑催化的烯炔与末端炔烃的 [2+2+2] 环加成反应。推定的机制涉及初始配体交换,然后是氧化环化、烯烃插入和还原消除过程。氧化环化被证明是完全化学转化的速率和选择性决定步骤,其中末端炔烃上的 R 取代基对反应选择性具有显着影响。当 R 是给电子基团(OMe 和 Me)时,邻位-取代的三环萘氢呋喃 ( P1 ) 预计将占主导地位;相反,当R为吸电子基团(NO 2、CF 3和CN)时,间位取代化合物P2成为主要产物。选择性的计算预测与实验产品比率非常一致。P1和P2的决速步骤的自由能垒为∼22.3–23.6 kcal mol –1,这与他们在 313 K 下 0.5 小时后的实验产率为 ~79-92% 非常吻合。结果还准确地再现了实验观察到的区域、化学和对映选择性,空间位阻以及底物和配体的电子特性显着影响反应速率和选择性。还详细探讨和讨论了计算方法的影响。

























 京公网安备 11010802027423号
京公网安备 11010802027423号