当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Diazines on graphene: adsorption, structural variances and electronic states
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2022-11-21 , DOI: 10.1039/d2cp05096j Oksana I Grinevich 1 , Victor V Volkov 2 , Aleksey K Buryak 1
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2022-11-21 , DOI: 10.1039/d2cp05096j Oksana I Grinevich 1 , Victor V Volkov 2 , Aleksey K Buryak 1
Affiliation
|
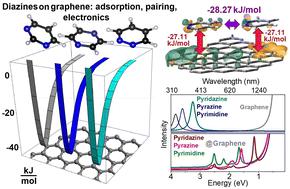
We conduct quantum studies of adsorption of diazine heterocycles on graphene to discuss experimental thermodynamics of gas-phase adsorption of pyridazine, pyrimidine and pyrazine on graphitized thermal carbon black, as reported previously. Using Born–Oppenheimer molecular dynamics and density functional studies, we characterize structural and electronic tendencies of the heterocycles on graphene. The theoretical studies predict the adsorption of pyridazine, pyrazine and pyrimidine to cause electronic perturbations of dipole, quadrupole and mixed spatial characters, respectively, resulting in a red shift of the electronic components of the heterocycles to modulate graphene electronics upon admixing of diazine orbital components with the πz states of the substrate. Investigating the thermodynamics of adsorption further involves calculating Henry's constant with the expression of the uniform surface limit: using experimental data, we estimate binding energies and force derivatives with respect to the surface normal. The extracted association energies agree with the results of Lennard-Jones potential calculations. Together, the reported pyridazine anomalous retention required the association force constant to be lower compared with values for the other diazines. Exploring energies of intermolecular relations, we ascribe the pyridazine anomalous retention to possibility of the formation of pyridazine dimers: when on the surface, only for pyridazine, the computed benefit of pairing is larger than the energy of molecular association with graphene.
中文翻译:

石墨烯上的二嗪:吸附、结构变化和电子态
我们对二嗪杂环在石墨烯上的吸附进行了量子研究,以讨论哒嗪、嘧啶和吡嗪在石墨化热炭黑上气相吸附的实验热力学,如前所述。使用 Born-Oppenheimer 分子动力学和密度泛函研究,我们表征了石墨烯上杂环的结构和电子趋势。理论研究预测哒嗪、吡嗪和嘧啶的吸附分别引起偶极子、四极子和混合空间特征的电子扰动,导致杂环电子组分的红移,从而在二嗪轨道组分与π z衬底的状态。研究吸附的热力学进一步涉及用均匀表面极限的表达式计算亨利常数:使用实验数据,我们估计结合能和相对于表面法线的力导数。提取的缔合能与 Lennard-Jones 势计算的结果一致。总之,所报道的哒嗪异常保留要求缔合力常数低于其他二嗪的值。探索分子间关系的能量,我们将哒嗪异常保留归因于哒嗪二聚体形成的可能性:当在表面上时,仅对于哒嗪,配对的计算收益大于分子与石墨烯缔合的能量。
更新日期:2022-11-21
中文翻译:
石墨烯上的二嗪:吸附、结构变化和电子态
我们对二嗪杂环在石墨烯上的吸附进行了量子研究,以讨论哒嗪、嘧啶和吡嗪在石墨化热炭黑上气相吸附的实验热力学,如前所述。使用 Born-Oppenheimer 分子动力学和密度泛函研究,我们表征了石墨烯上杂环的结构和电子趋势。理论研究预测哒嗪、吡嗪和嘧啶的吸附分别引起偶极子、四极子和混合空间特征的电子扰动,导致杂环电子组分的红移,从而在二嗪轨道组分与π z衬底的状态。研究吸附的热力学进一步涉及用均匀表面极限的表达式计算亨利常数:使用实验数据,我们估计结合能和相对于表面法线的力导数。提取的缔合能与 Lennard-Jones 势计算的结果一致。总之,所报道的哒嗪异常保留要求缔合力常数低于其他二嗪的值。探索分子间关系的能量,我们将哒嗪异常保留归因于哒嗪二聚体形成的可能性:当在表面上时,仅对于哒嗪,配对的计算收益大于分子与石墨烯缔合的能量。


























 京公网安备 11010802027423号
京公网安备 11010802027423号