当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Generalization of ETS-NOCV and ALMO-COVP Energy Decomposition Analysis to Connect Any Two Electronic States and Comparative Assessment
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-11-18 , DOI: 10.1021/acs.jctc.2c00901 Hengyuan Shen 1 , Zhenling Wang 1 , Martin Head-Gordon 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2022-11-18 , DOI: 10.1021/acs.jctc.2c00901 Hengyuan Shen 1 , Zhenling Wang 1 , Martin Head-Gordon 1
Affiliation
|
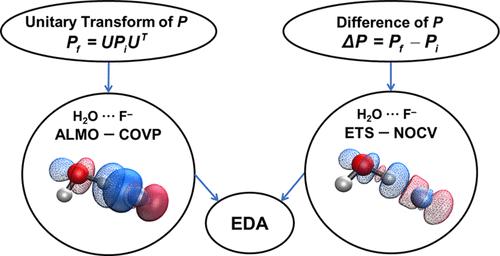
Energy decomposition analysis (EDA) is a useful tool for obtaining chemically meaningful insights into molecular interactions. The extended transition-state method with natural orbitals for chemical valence (ETS-NOCV) and the absolutely localized molecular orbital-based method with complementary occupied-virtual pairs (ALMO-COVP) are two successful EDA schemes. Working within ground-state generalized Kohn–Sham density functional theory (DFT), we extend these methods to perform EDA between any two electronic states that can be connected by a unitary transformation of density matrices. A direct proof that the NOCV eigenvalues are symmetric pairs is given, and we also prove that the charge and energy difference defined by ALMO are invariant under certain orbital rotations, allowing us to define COVPs. We point out that ETS is actually a 1-point quadrature to obtain the effective Fock matrix, and though it is reasonably accurate, it can be systematically further improved by adding more quadrature points. We explain why the calculated amount of transferred charge measured by ALMO-COVP is typically much smaller than that of ETS-NOCV and explain why the ALMO-COVP values should be preferred. While the two schemes are independent, ETS-NOCV and ALMO-COVP in fact give a very similar chemical picture for a variety of chemical interactions, including H–H+, the transition structure for the Diels–Alder reaction between ethene and butadiene, and two hydrogen-bonded complexes, H2O···F– and H2O···HF.
中文翻译:

ETS-NOCV 和 ALMO-COVP 能量分解分析的推广以连接任何两个电子状态和比较评估
能量分解分析 (EDA) 是一种有用的工具,可用于获得对分子相互作用具有化学意义的见解。具有化学价自然轨道的扩展过渡态方法 (ETS-NOCV) 和具有互补占据-虚对的基于绝对局域化分子轨道的方法 (ALMO-COVP) 是两个成功的 EDA 方案。在基态广义 Kohn-Sham 密度泛函理论 (DFT) 中工作,我们扩展了这些方法以在可以通过密度矩阵的酉变换连接的任何两个电子态之间执行 EDA。给出了 NOCV 特征值是对称对的直接证明,我们还证明了 ALMO 定义的电荷和能量差在某些轨道旋转下是不变的,从而允许我们定义 COVP。我们指出,ETS 实际上是一个 1 点积分以获得有效的 Fock 矩阵,虽然它相当准确,但可以通过添加更多积分点来系统地进一步改进。我们解释了为什么 ALMO-COVP 测量的计算转移电荷量通常比 ETS-NOCV 小得多,并解释了为什么应首选 ALMO-COVP 值。虽然这两个方案是独立的,但 ETS-NOCV 和 ALMO-COVP 实际上为各种化学相互作用给出了非常相似的化学图,包括 H–H 我们解释了为什么 ALMO-COVP 测量的计算转移电荷量通常比 ETS-NOCV 小得多,并解释了为什么应首选 ALMO-COVP 值。虽然这两个方案是独立的,但 ETS-NOCV 和 ALMO-COVP 实际上为各种化学相互作用给出了非常相似的化学图,包括 H–H 我们解释了为什么 ALMO-COVP 测量的计算转移电荷量通常比 ETS-NOCV 小得多,并解释了为什么应首选 ALMO-COVP 值。虽然这两个方案是独立的,但 ETS-NOCV 和 ALMO-COVP 实际上为各种化学相互作用给出了非常相似的化学图,包括 H–H+,乙烯和丁二烯之间 Diels-Alder 反应的过渡结构,以及两个氢键配合物,H 2 O···F –和 H 2 O···HF。
更新日期:2022-11-18
中文翻译:
ETS-NOCV 和 ALMO-COVP 能量分解分析的推广以连接任何两个电子状态和比较评估
能量分解分析 (EDA) 是一种有用的工具,可用于获得对分子相互作用具有化学意义的见解。具有化学价自然轨道的扩展过渡态方法 (ETS-NOCV) 和具有互补占据-虚对的基于绝对局域化分子轨道的方法 (ALMO-COVP) 是两个成功的 EDA 方案。在基态广义 Kohn-Sham 密度泛函理论 (DFT) 中工作,我们扩展了这些方法以在可以通过密度矩阵的酉变换连接的任何两个电子态之间执行 EDA。给出了 NOCV 特征值是对称对的直接证明,我们还证明了 ALMO 定义的电荷和能量差在某些轨道旋转下是不变的,从而允许我们定义 COVP。我们指出,ETS 实际上是一个 1 点积分以获得有效的 Fock 矩阵,虽然它相当准确,但可以通过添加更多积分点来系统地进一步改进。我们解释了为什么 ALMO-COVP 测量的计算转移电荷量通常比 ETS-NOCV 小得多,并解释了为什么应首选 ALMO-COVP 值。虽然这两个方案是独立的,但 ETS-NOCV 和 ALMO-COVP 实际上为各种化学相互作用给出了非常相似的化学图,包括 H–H 我们解释了为什么 ALMO-COVP 测量的计算转移电荷量通常比 ETS-NOCV 小得多,并解释了为什么应首选 ALMO-COVP 值。虽然这两个方案是独立的,但 ETS-NOCV 和 ALMO-COVP 实际上为各种化学相互作用给出了非常相似的化学图,包括 H–H 我们解释了为什么 ALMO-COVP 测量的计算转移电荷量通常比 ETS-NOCV 小得多,并解释了为什么应首选 ALMO-COVP 值。虽然这两个方案是独立的,但 ETS-NOCV 和 ALMO-COVP 实际上为各种化学相互作用给出了非常相似的化学图,包括 H–H+,乙烯和丁二烯之间 Diels-Alder 反应的过渡结构,以及两个氢键配合物,H 2 O···F –和 H 2 O···HF。

























 京公网安备 11010802027423号
京公网安备 11010802027423号