Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2022-11-17 , DOI: 10.1016/j.csbj.2022.11.032 Rahul Kaushik 1 , Kam Y J Zhang 1
|
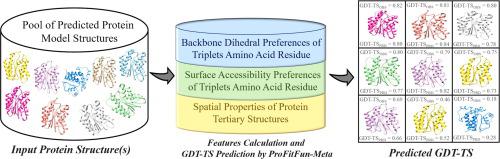
The structural information of a protein is pivotal to comprehend its functions, protein-protein and protein-ligand interactions. There is a widening gap between the number of known protein sequences and that of experimentally determined structures. The protein structure prediction has emerged as an efficient alternative to deliver the reliable structural information of proteins. However, it remains a challenge to identify the best model among the many predicted by one or a few structure prediction methods. Here we report ProFitFun-Meta, a neural network based pure single model scoring method for assessing the quality of predicted model structures by an effective combination structural information of various backbone dihedral angle and residue surface accessibility preferences of amino acid residues with other spatial properties of protein structures. The performance of ProFitFun-Meta was validated and benchmarked against current state-of-the-art methods on the extensive datasets, comprising a Test Dataset (n = 26,604), an External Dataset (n = 40,000), and CASP14 Dataset (n = 1200). The comprehensive performance evaluation of ProFitFun-Meta demonstrated its reliability and efficiency in terms of Spearman’s (ρ) and Pearson’s (r) correlation coefficients, GDT-TS loss (g), and absolute loss (d). An improved performance over the current state-of-the-art methods and leading performers of CASP14 experiment in quality assessment category demonstrated its potential to become an integral component of computational pipelines for protein modeling and design. The minimal dependencies, high computational efficiency, and portability to various Linux and Windows OS provide an additional edge to ProFitFun-Meta for its easy implementation and applications in various regimes of computational protein folding.
中文翻译:
一种用于识别类天然模型结构的综合蛋白质结构适应度评分方法
蛋白质的结构信息对于理解其功能、蛋白质-蛋白质和蛋白质-配体相互作用至关重要。已知蛋白质序列的数量与实验确定的结构数量之间存在越来越大的差距。蛋白质结构预测已成为提供可靠的蛋白质结构信息的有效替代方法。然而,在由一种或几种结构预测方法预测的众多模型中确定最佳模型仍然是一个挑战。这里我们报告ProFitFun-Meta,一种基于神经网络的纯单一模型评分方法,通过氨基酸残基的各种主链二面角和残基表面可及性偏好与蛋白质结构的其他空间特性的有效组合结构信息来评估预测模型结构的质量。ProFitFun-Meta 的性能在广泛的数据集上根据当前最先进的方法进行了验证和基准测试,包括测试数据集 (n = 26,604)、外部数据集 (n = 40,000) 和 CASP14 数据集 (n = 1200)。ProFitFun-Meta 的综合性能评估在斯皮尔曼 (ρ) 和皮尔逊 (r) 相关系数、GDT-TS 损失 (g) 和绝对损失 (d) 方面证明了其可靠性和效率。与当前最先进的方法相比,CASP14 实验在质量评估类别中的领先表现有所改进,证明了它有可能成为蛋白质建模和设计计算管道的一个组成部分。最小的依赖性、高计算效率和对各种 Linux 和 Windows 操作系统的可移植性为 ProFitFun-Meta 提供了额外的优势,因为它可以在各种计算蛋白质折叠机制中轻松实施和应用。

























 京公网安备 11010802027423号
京公网安备 11010802027423号